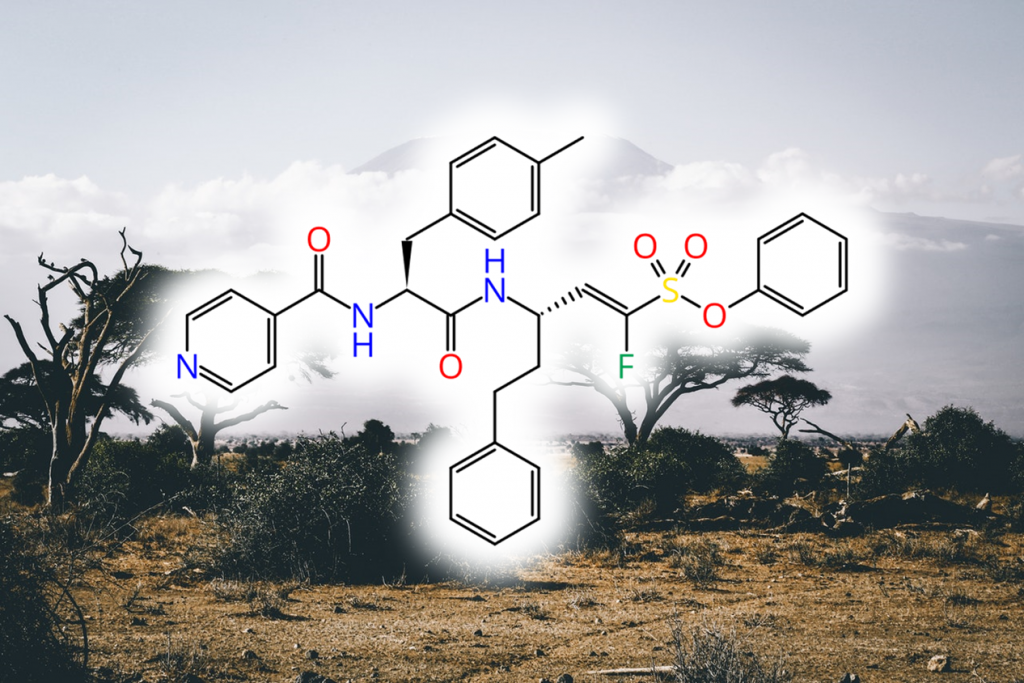

Human African Trypanosomiasis (HAT, African Sleeping Sickness) is a fatal, neglected tropical disease caused by the parasites Trypanosoma brucei. Most available drugs for treatment of the disease lack efficiency and have severe side effects. α-halovinylsulfones as covalent reversible inhibitors of the parasitic cysteine protease “rhodesain” have proven to be promising novel drug candidates.
Here, the team around Tanja focused on optimizing α-fluorovinylsulfones and -sulfonates for rhodesain inhibition using molecular modeling approaches. This resulted in highly potent and selective inhibitors with single-digit nanomolar affinity. The researchers further investigated the binding modes experimentally via MS experiments, indicating that the compounds are covalent-reversible, slow-tight binders. The different inhibition mechanisms of fluorinated and non-fluorinated compounds (reversible vs. irreversible) were investigated by QM/MM calculations and MD simulations.
In vivo studies revealed a favorable metabolism and biodistribution compared to non-optimized rhodesain inhibitors. Furthermore, they observed an anti-trypanosomal activity in the nanomolar range for inhibitors with an N-terminal 2,3-dihydrobenzo[b][1,4]dioxine group and a 4-Me-Phe residue in P2.