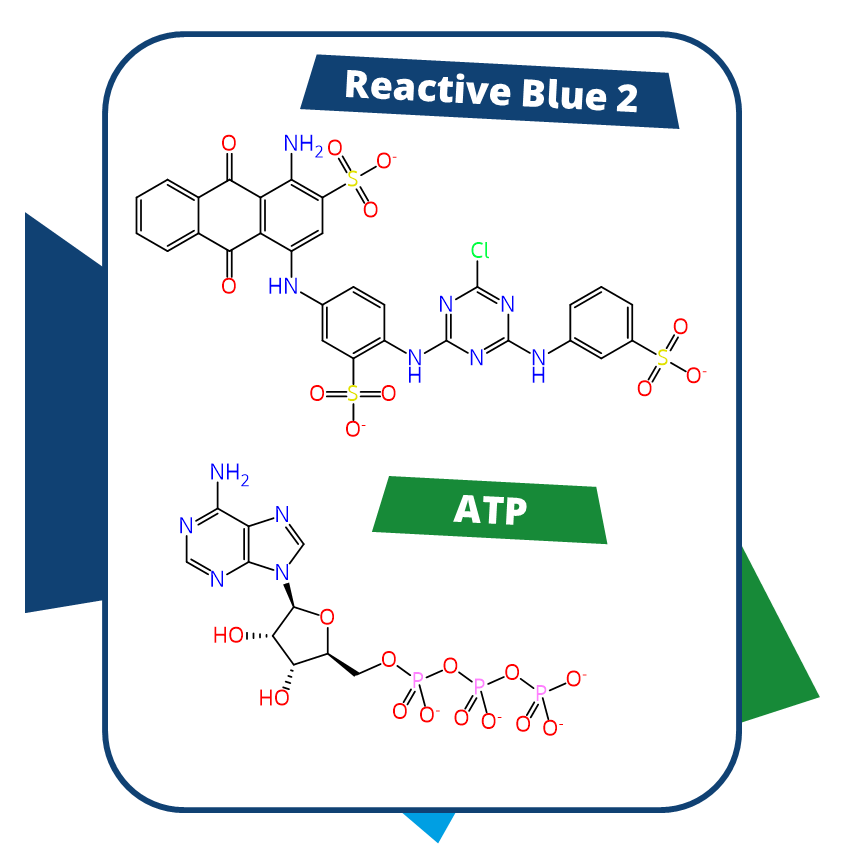
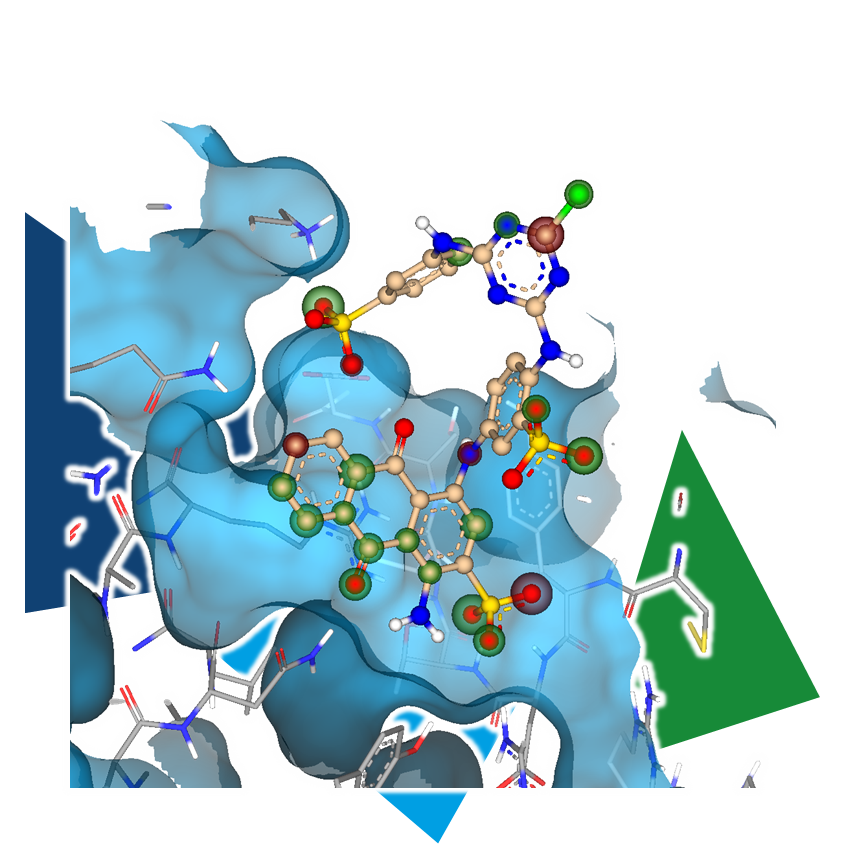
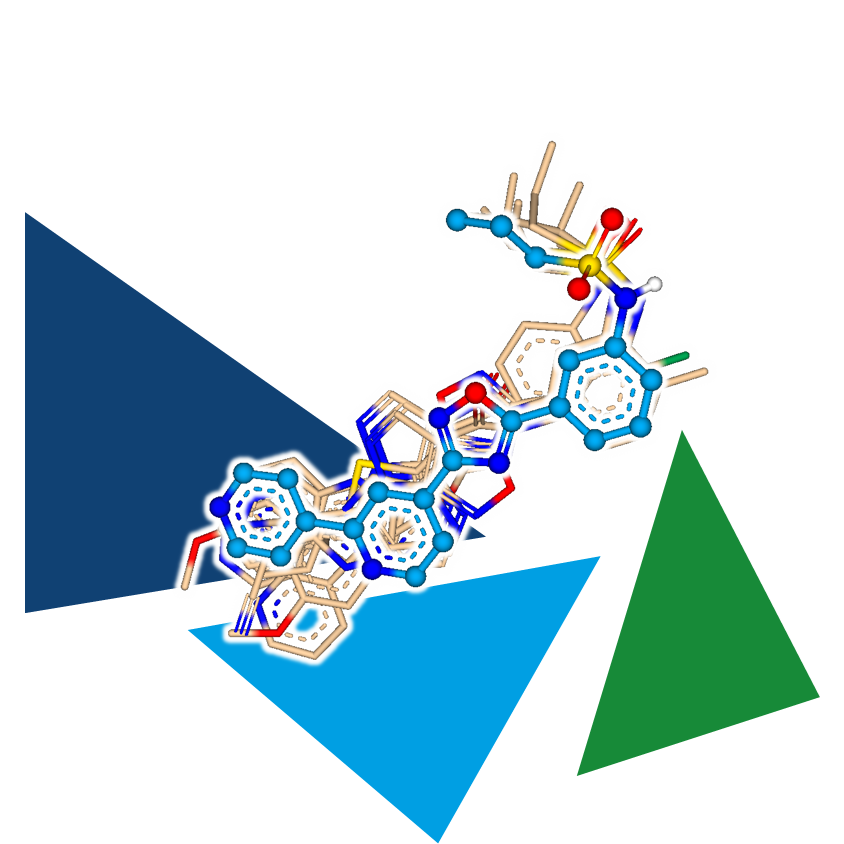
Virtual screening does not necessarily require a target structure. Ligand-based methods utilize a known binder (in our case, the tool compound) to align it with a molecule set and identify compounds that display 3D similarity to the ligand structure.
It is worth noting that information about interactions, including potential clashes within the binding site, is not considered in this approach. While this might seem like a significant loss of information, it is important to remember that structure-based approaches often rely on a single binding mode hypothesis, which is typically generated through docking. Multifunctional tool compounds may exhibit multiple potential binding modes, and without a series of compounds, there may not be enough data to establish coherent SARs. This inherent fuzziness of ligand-based methods can sometimes be an advantage, still leading to the identification of active hits.
BioSolveIT software for 3D LBDD:
- SeeSAR's Similarity Scanner Mode: Can perform ligand-based virtual screening using a tool compound as query. Pharmacophore constraints can be applied to improve and fine-tune the results.
Command-line tools for 3D LBDD: