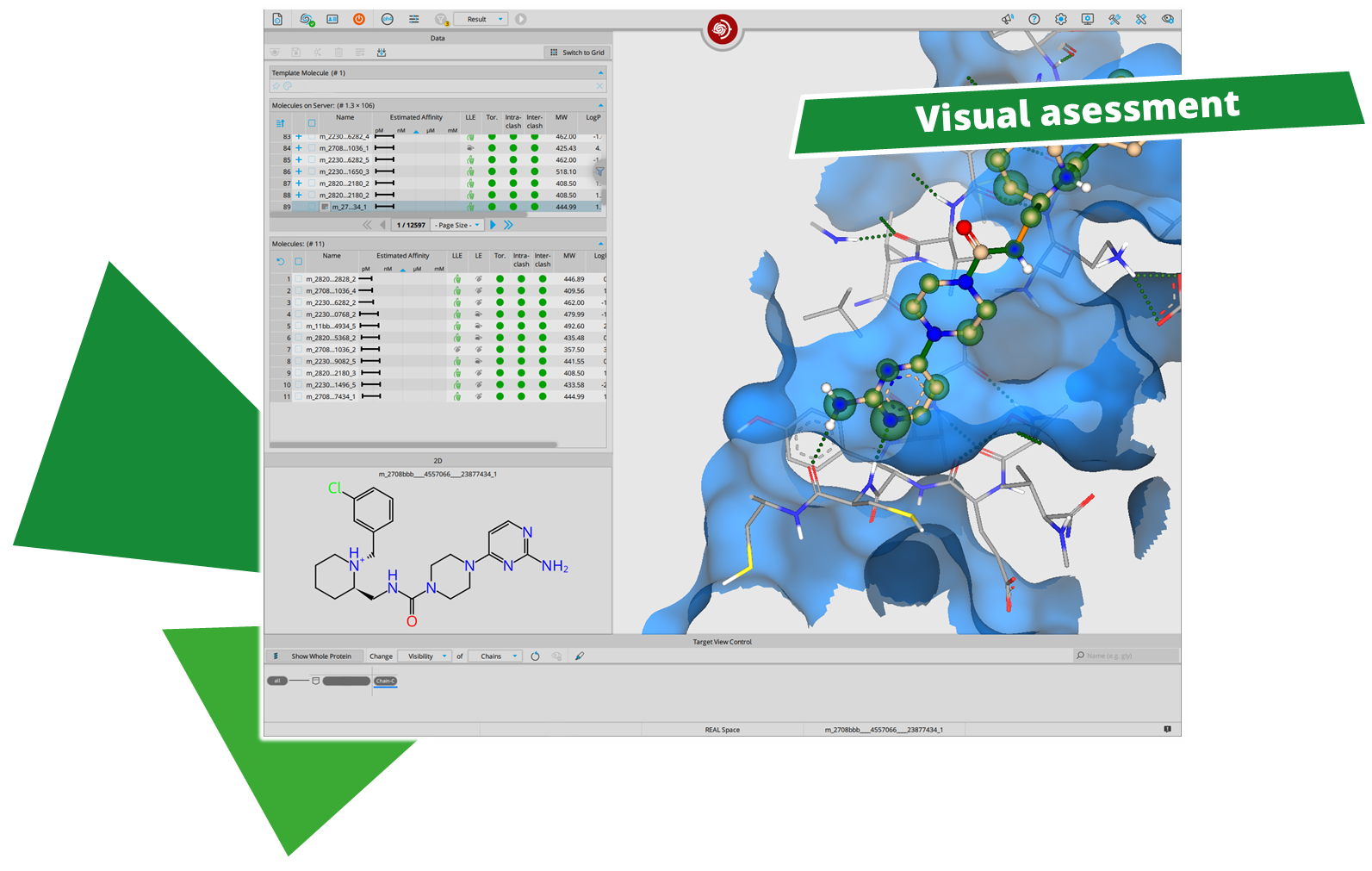
After performing a search with infiniSee your results will be presented in a table. The column "Source" tells you the origin of the Chemical Space that contains your solution; the ID of the respective result molecule is shown in the "Name" column.
Compounds can be ordered by sending a quote request to the compound vendor with the following information:
Requested structures in SMILES or SD format, Compound ID (concatenated), and amount requested.
For compounds from Ambinter's AMBrosia Space, send your request to
ambrosia@greenpharma.com.
For compounds from eMolecule's eXplore Space, send your request to
explore@emolecules.com.
For compounds from Enamine's REAL Space, send your request to
libraries@enamine.net.
For compounds from WuXi's GalaXi Space please send your request to
contact@labnetwork.com
For compounds from OTAVA's CHEMriya Space please send your request to
info@otava.ca.
For compounds from Chemspace's Freedom Space please send your request to
sales@chem-space.com.
 SeeSAR
SeeSAR