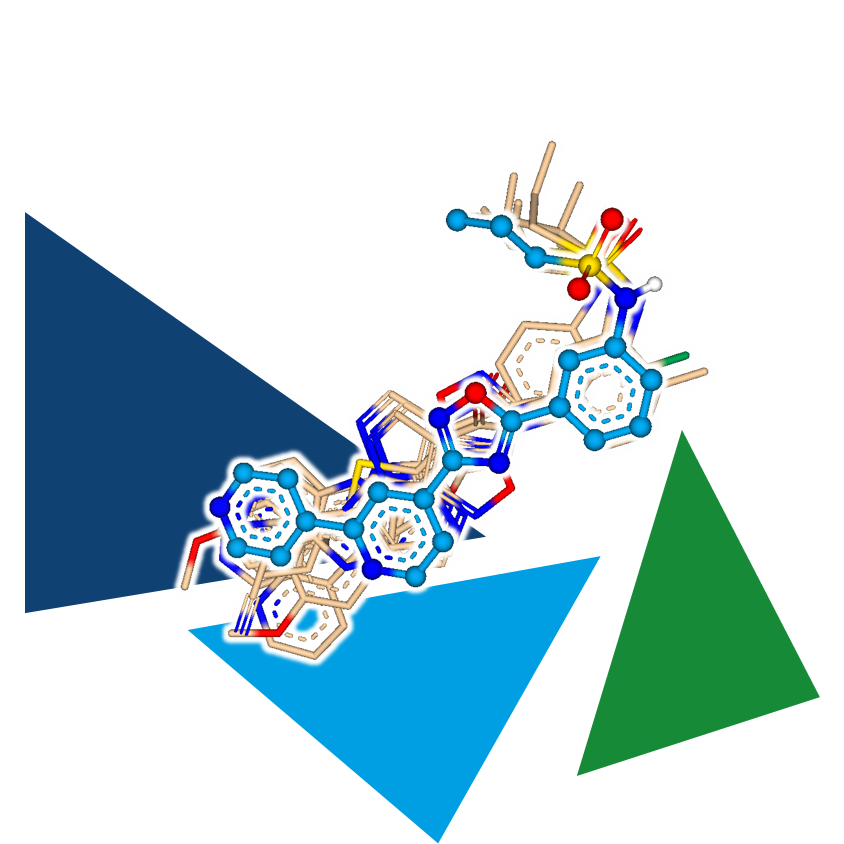
Hit to lead campaigns can also profit from 3D ligand-based approaches.
Here, a template molecule is used on a compound library. The entries are aligned with the aim to match similar molecular features and fit the shape of the query. Compounds displaying similar arrangement of functionalities have a higher propbability of displaying similar biological activities at the target.
BioSolveIT software for 3D LBDD:
- SeeSAR's Similarity Scanner Mode: Ligand-based virtual screening.
Command-line tools for 3D LBDD: