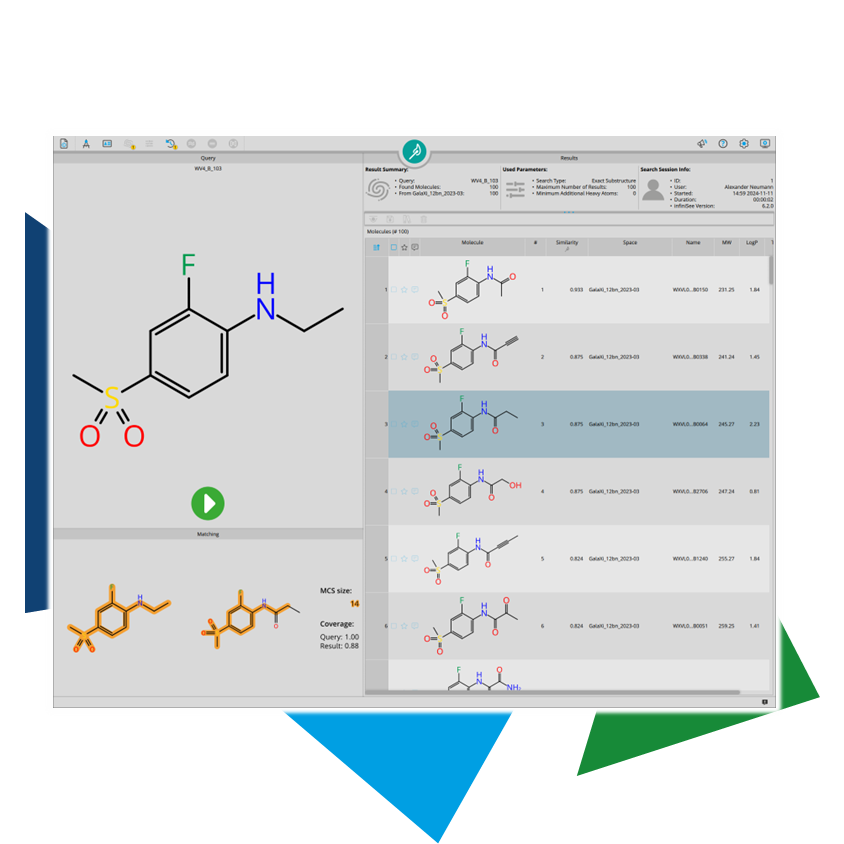
Fragments are defined by the
rule of three: no more than three hydrogen bond acceptors and donors each, no more than three rotatable bonds, a molecular weight of less than 300, and a logP no greater than 3. This ensures that the compound will not violate the rule of five during lead optimization when additional functionalities are added.
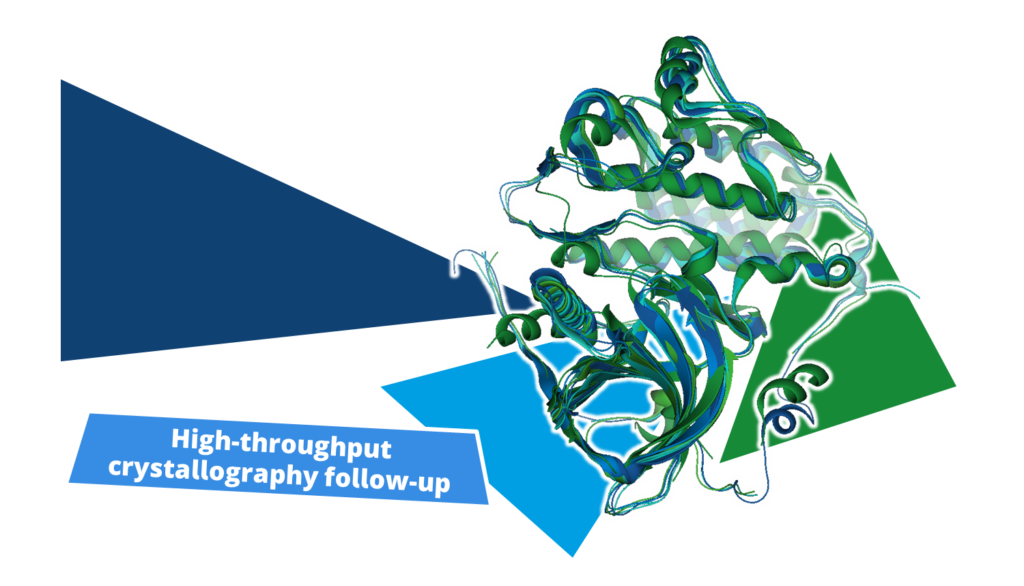
During a high-throughput crystallographic fragment screening, target structures are exposed to fragments in an attempt to crystallize complexes that contain a binder. With the right capacity, hundreds of crystal structures of a single target can be collected, each containing or or more fragments, sometimes even at different binding sites. This is where the decision-making process begins, determining which fragments are of particular interest and should be further optimized in the subsequent steps.