Virtual Screening: Your Drug Discovery Accelerator
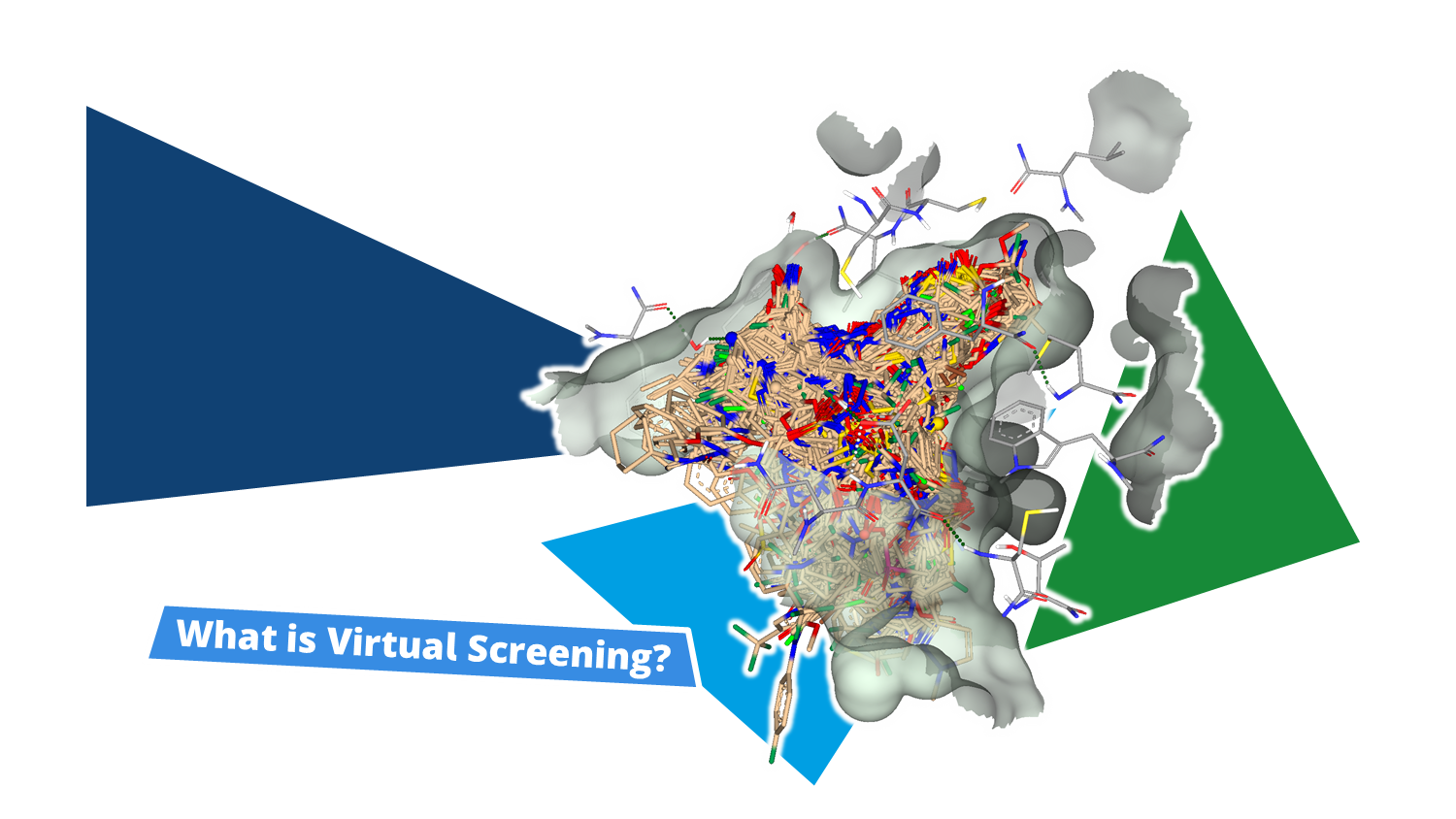
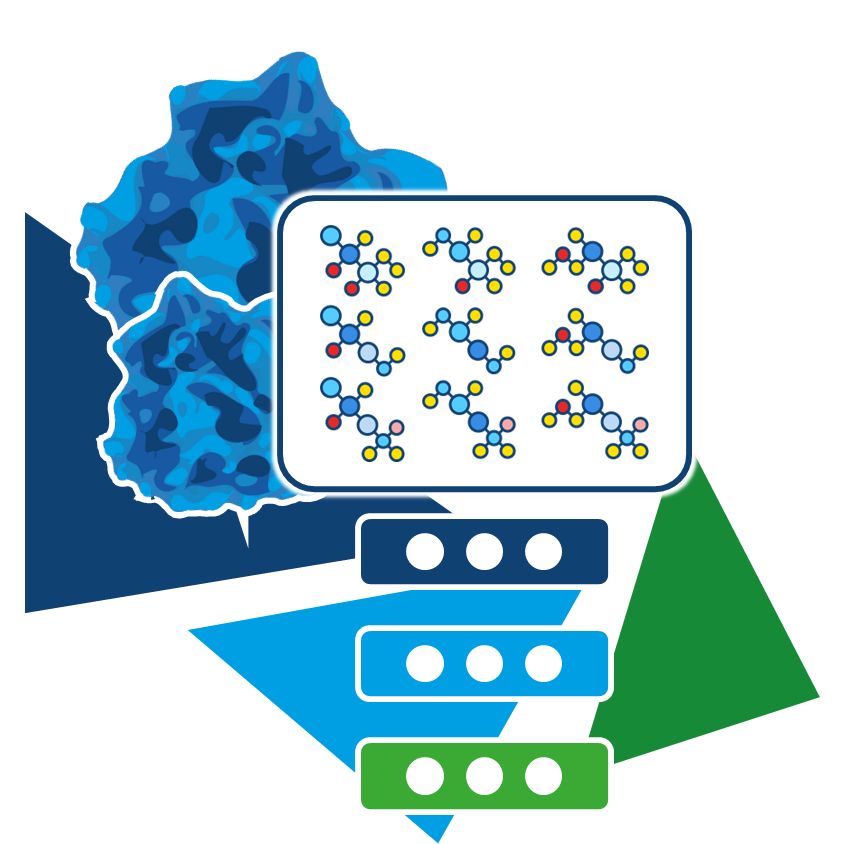
Virtual screening is a computational approach that predicts potential binders from large molecular datasets—often comprising millions, billions, or even trillions of compounds.
It mines for the most promising candidates for experimental validation and thus minimizes experimental costs and maximizes efficiency in the early phases of drug discovery.
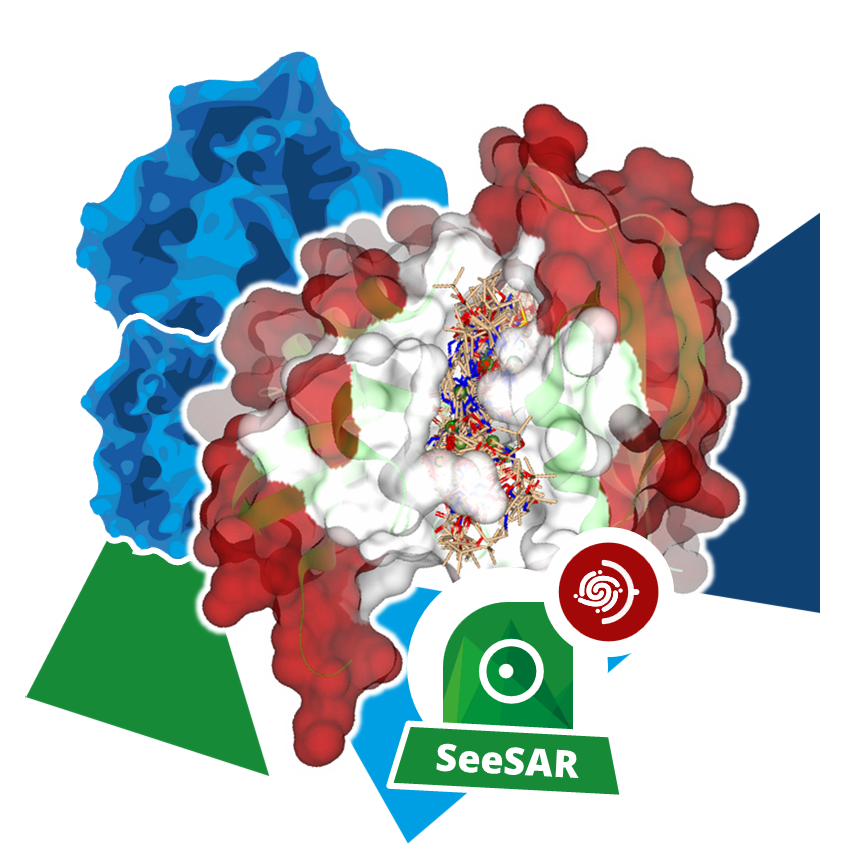
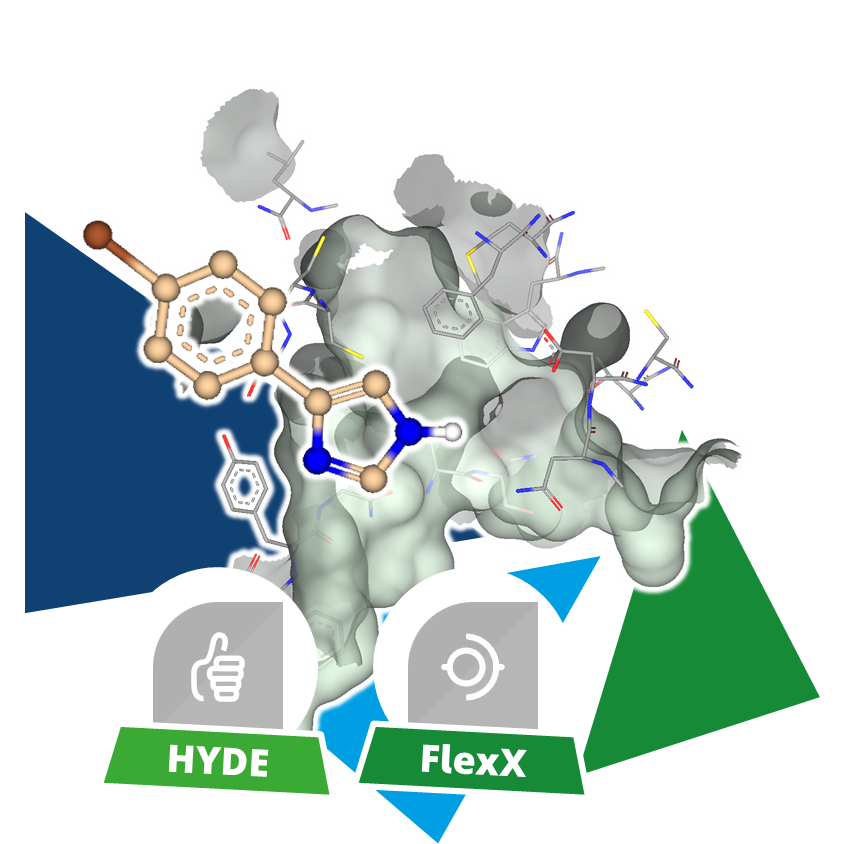
Given a set of compounds, binding poses for each molecule at a target's binding site are generated and assessed for their formed interactions, leading to a numerical score. By ranking each pose based on it's score, compound with increased likelihood to form high-quality interactions with the target enrich in the top ranks. This is applied in early stages of drug discovery to focus on the compounds most likely to display biological activity by binding to a the structure of interest. After visual assessment of the virtual screening results, a selection of chemically diverse compounds is made to be tested, reducing the required resources to only a fraction of evaluating every single compound from the molecule set.