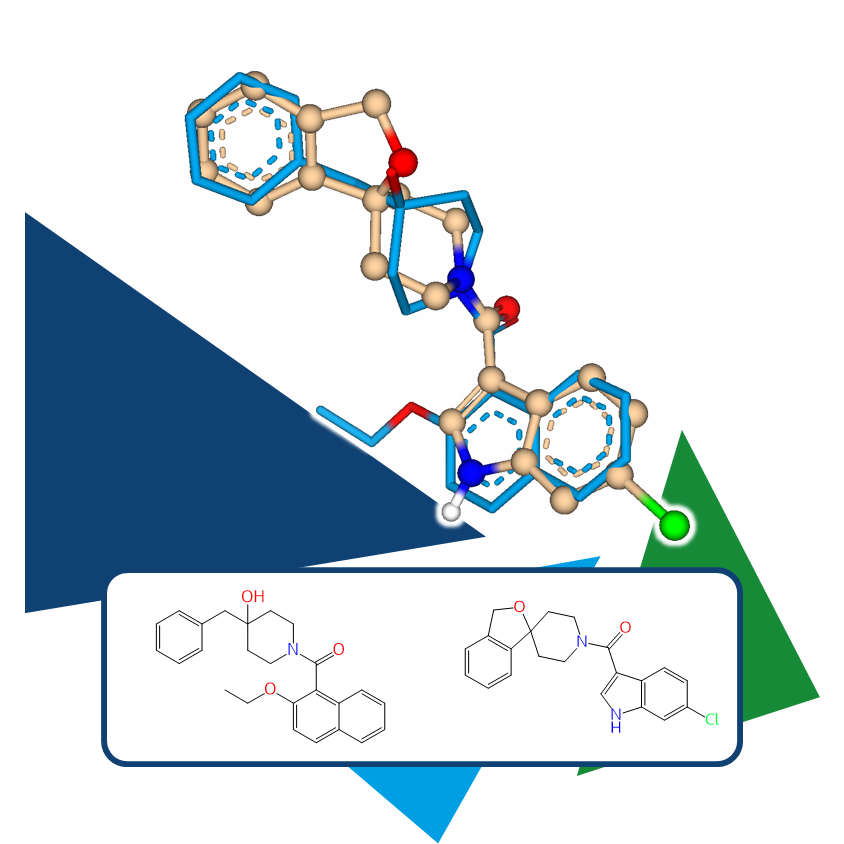
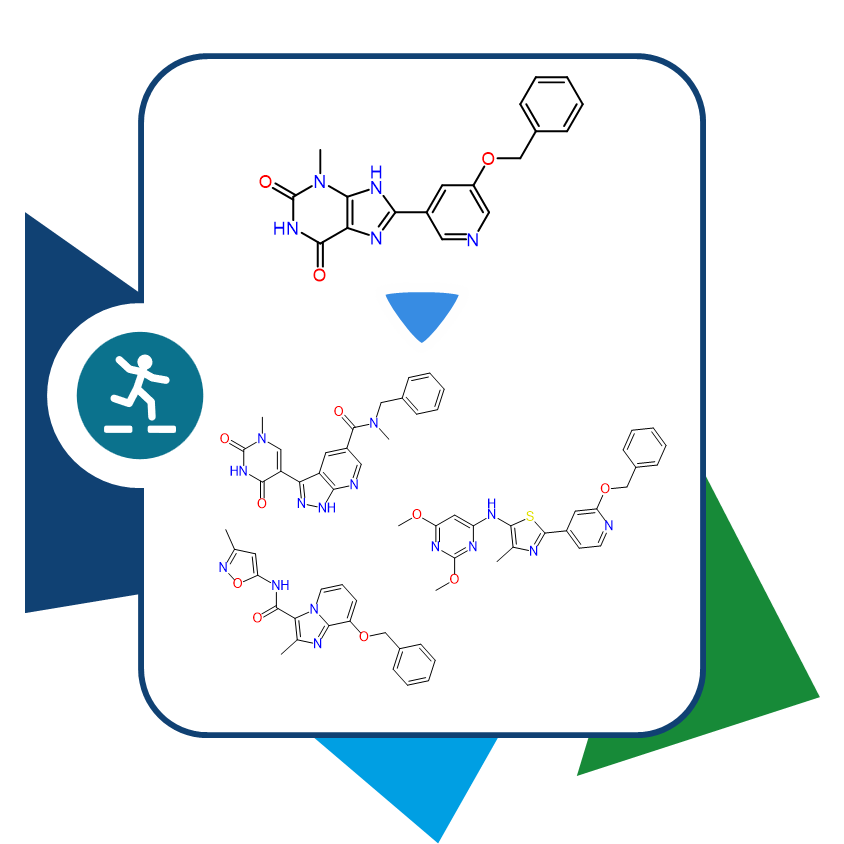
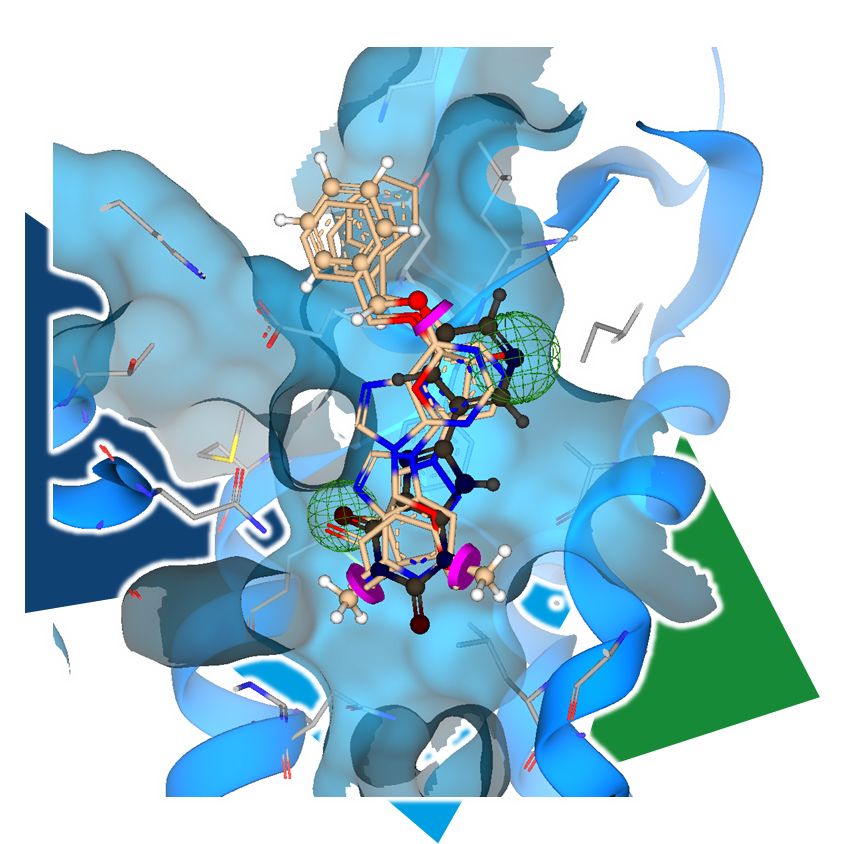
As an independent or complementary method, 3D molecule alignment can add the necessary refinement to results, enabling the identification of even more precisely similar pharmacophoric arrangements.
It is particularly important to incorporate key project insights into this process. For example, if multiple key features that define the pharmacophore are known, constraints can be applied to the template molecule to ensure that the resulting compounds maintain these functionalities in a 3D arrangement.
3D molecule alignment and superposition-focused virtual screening in the context of scaffold-based drug design can be achieved with following BioSolveIT platform:
- SeeSAR's Similarity Scanner Mode: Ligand-based virtual screening.
Command-line tools for 3D superpositioning: