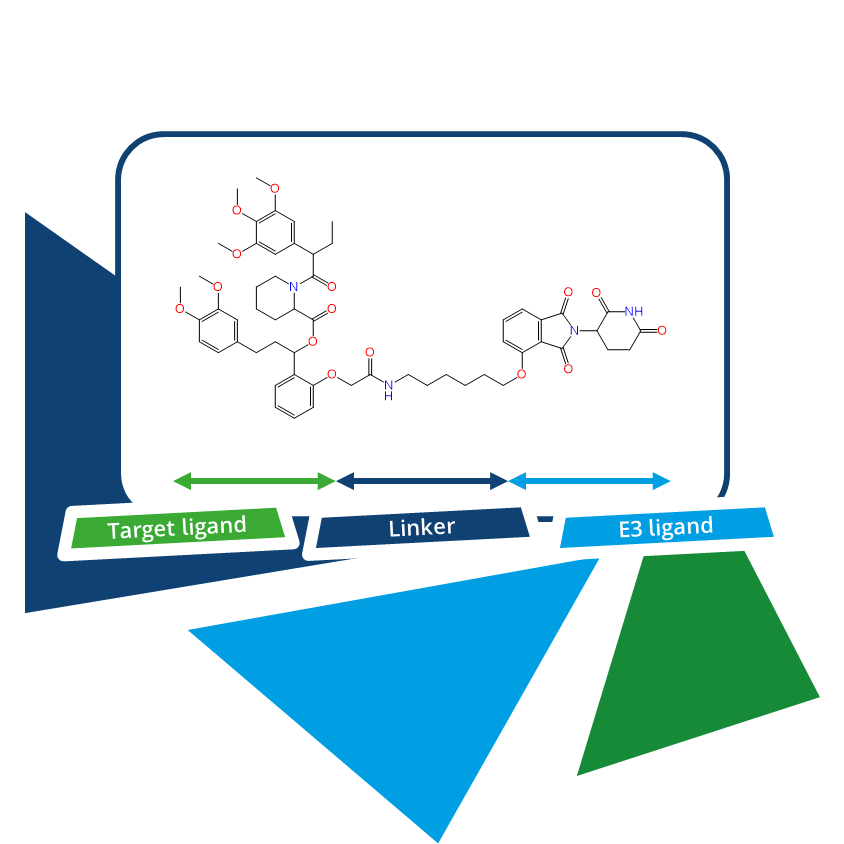
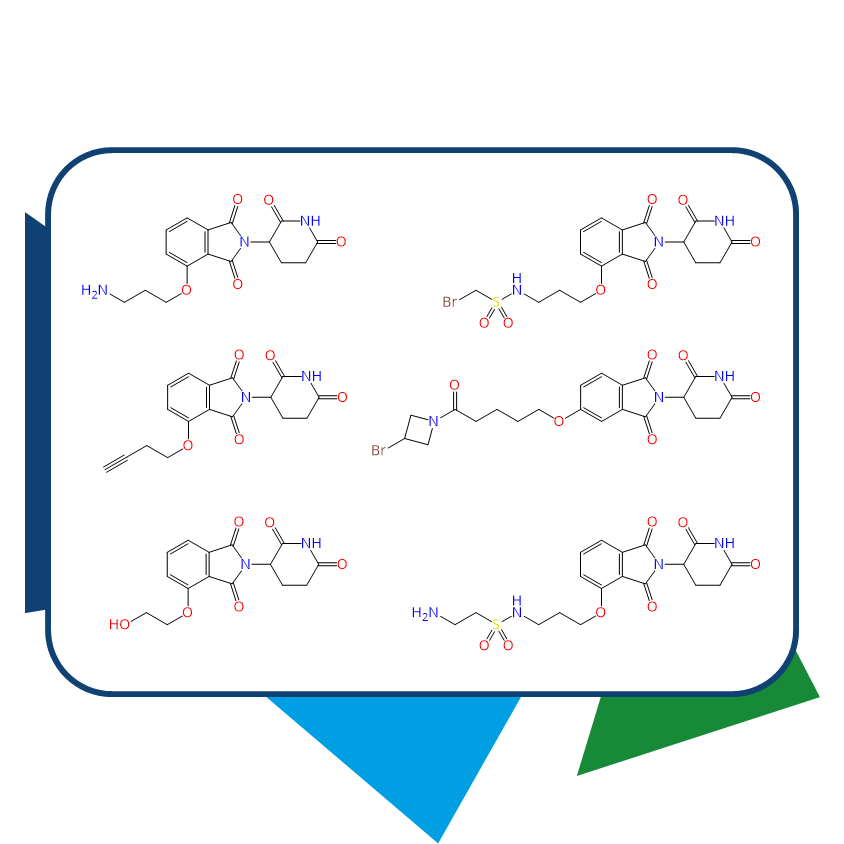
Chemical Spaces are Boutiques for PROTACs Design
Online compound catalogs rarely list more than 1,000 analogs of a ligase ligand. While this is not an insignificant number, most researchers would agree that it is all too common to encounter situations where certain building blocks are missing. Additionally, the external synthesis of desired building blocks can be quite costly.
Chemical Spaces address this issue by including all possible structures that can be synthesized from the available building blocks, exponentially expanding the range of options. For example, searching the eXplore database yields 1,339,094,441 structures containing lenalidomide as a substructure, 2,647,921,721 analogs for pomalidomide, and another 2,522,855,058 options for thalidomide.
The comparison between a thousand options and billions clearly highlights the potential that Chemical Spaces have in PROTAC design.