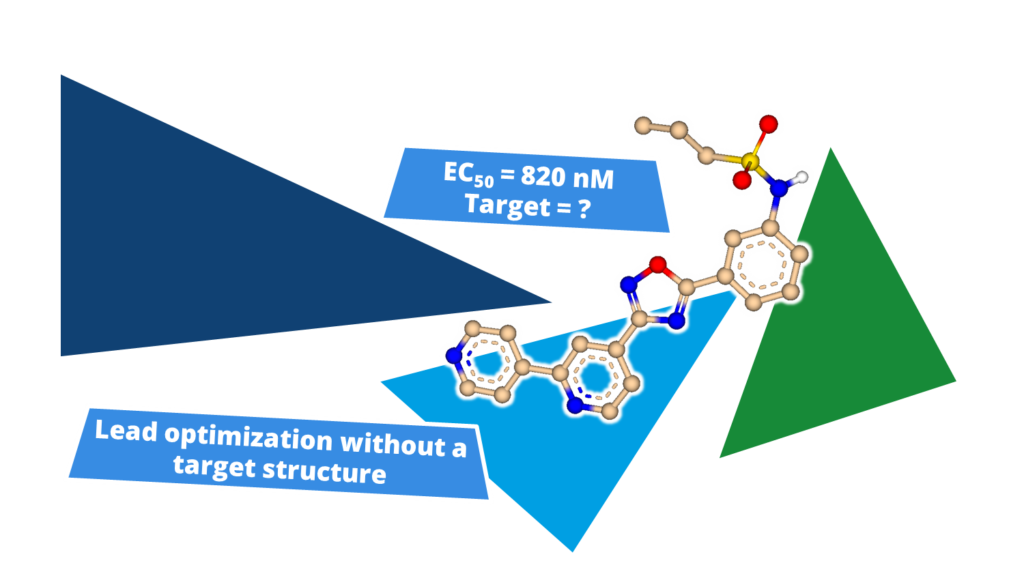
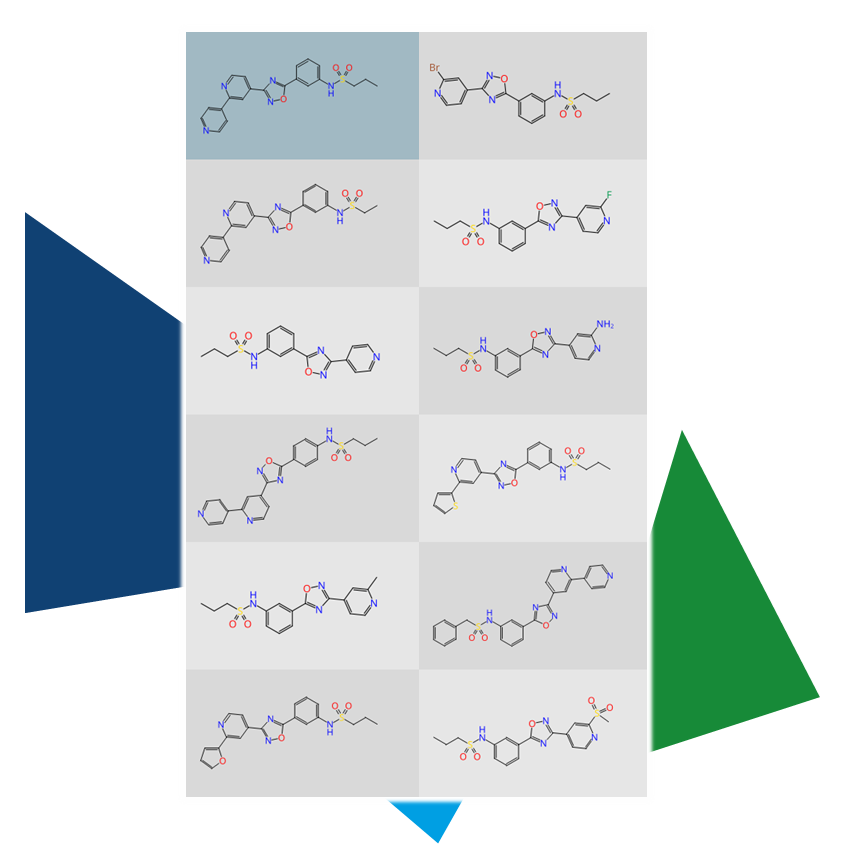
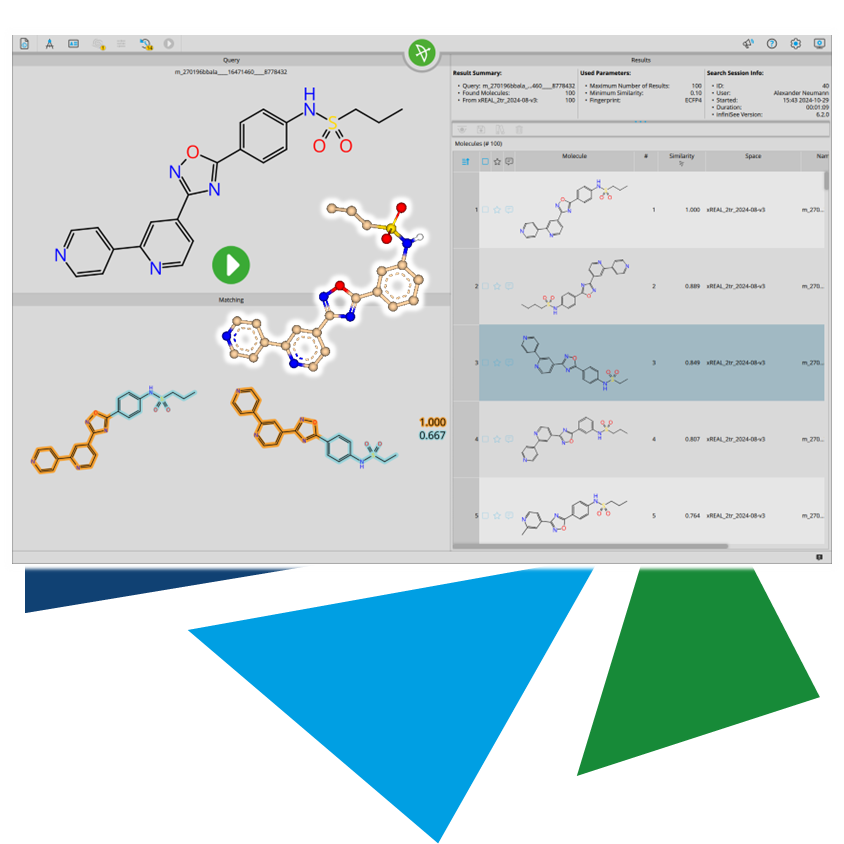
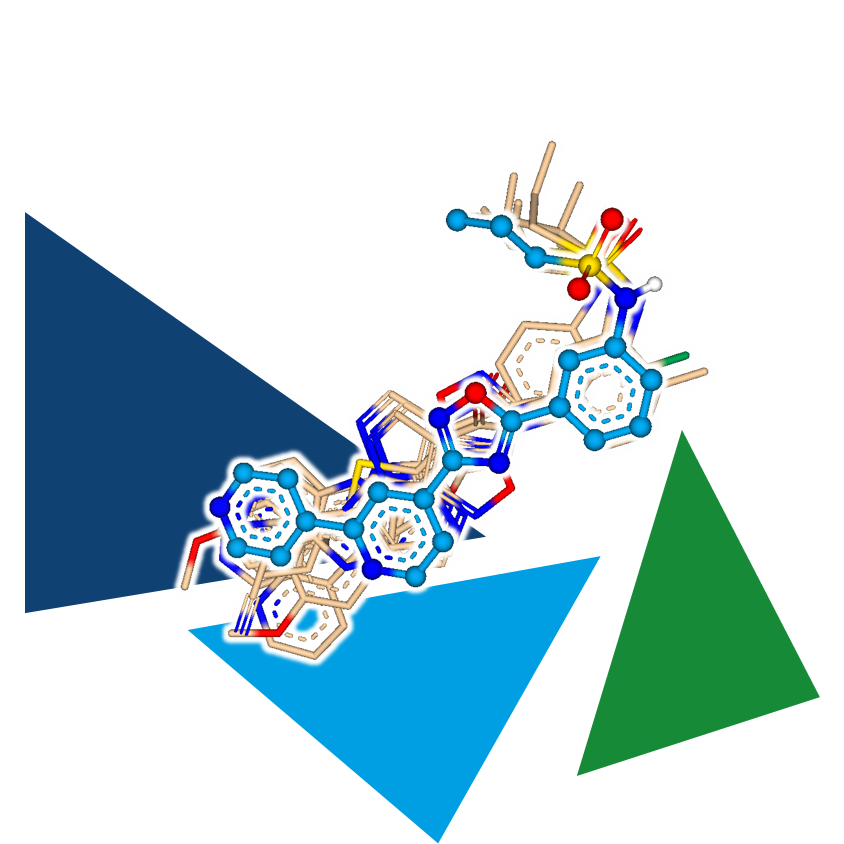
Furthermore, ligand-based lead optimization can benefit from 3D methods.
The additional dimension allows for spotting potential new scaffolds that would remain hidden in a two-dimensional approach. In particular, the combination of scaffold hopping (infiniSee's Scaffold Hopper Mode/FTrees) and 3D alignment opens up possibilities to match the shape of the template compound with entirely new structural classes through ring system rearrangement.
BioSolveIT software for 3D LBDD:
- SeeSAR's Similarity Scanner Mode: Ligand-based virtual screening.
Command-line tools for 3D LBDD: