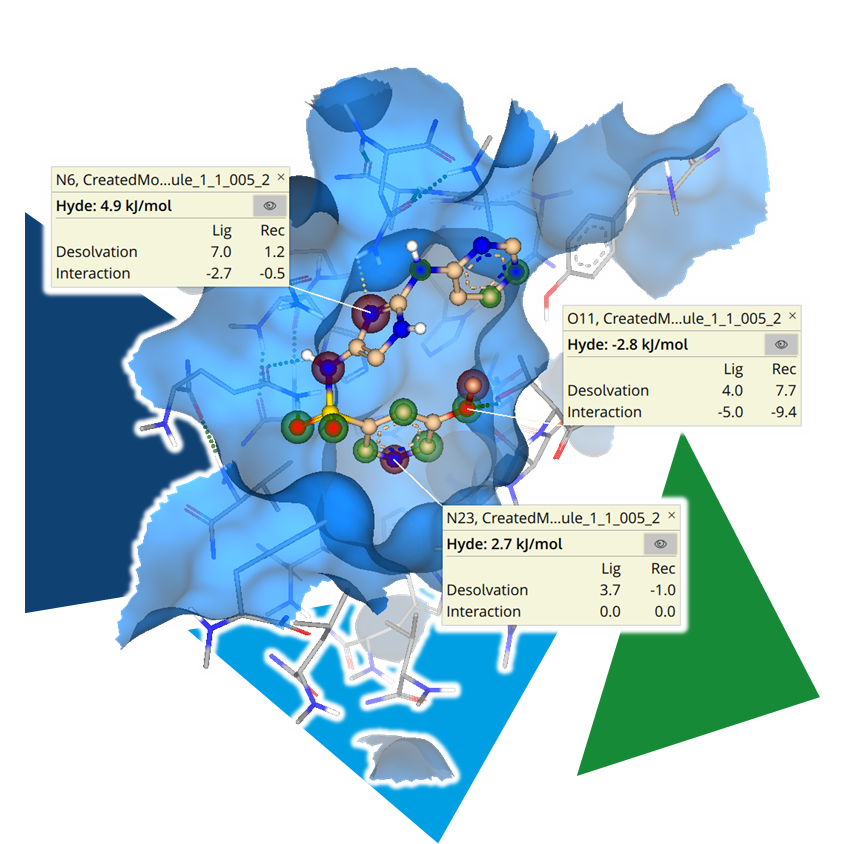
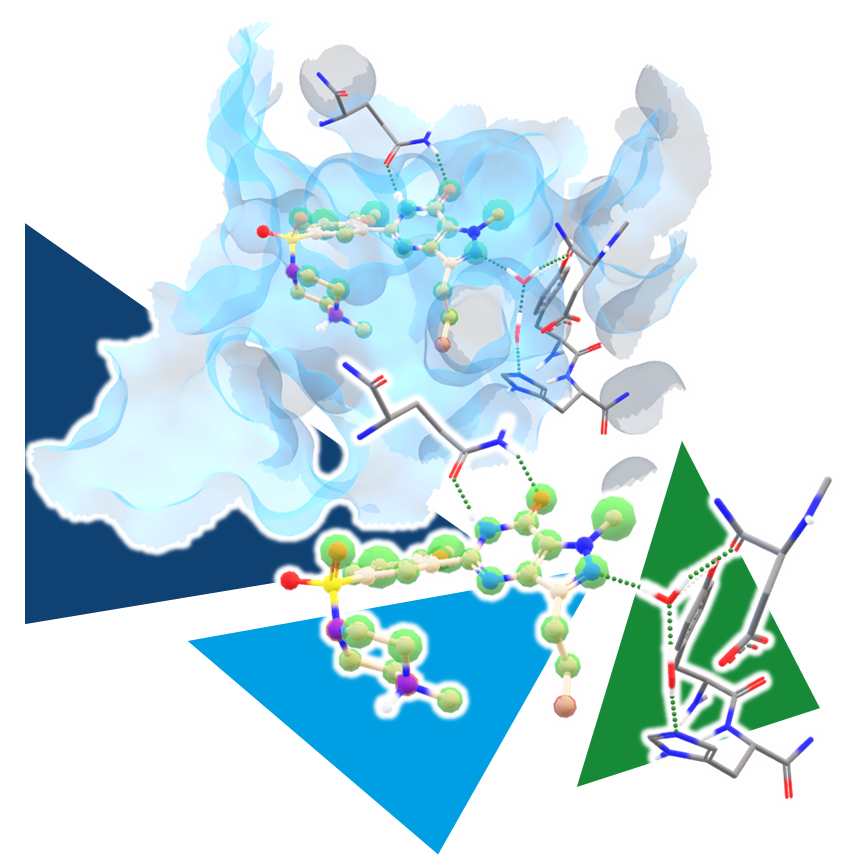
CROs for Drug Discovery: Partners for Research
CROs (
Contract Research Organizations) are companies specialized in independently conducting in-house R&D. They often have capabilities in synthesis, design, and testing, allowing them to serve as supportive or leading partners for pharmaceutical companies and other organizations that lack these capabilities. CROs assist in one or more projects, providing expertise to accelerate research processes and drug development.
A good CRO (Contract Research Organization) stands out by being multidisciplinary, offering its services as a comprehensive package where clients receive fully detailed information to advance their projects. However, it's also possible for clients to opt for specific services, such as the synthesis of a compound series, conducting virtual screenings, or structural elucidation of a target protein.
In all cases, a CRO should be versatile and well-equipped to provide the best results, ensuring flexibility to cater to diverse client needs.