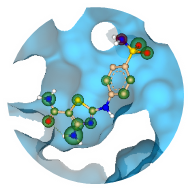
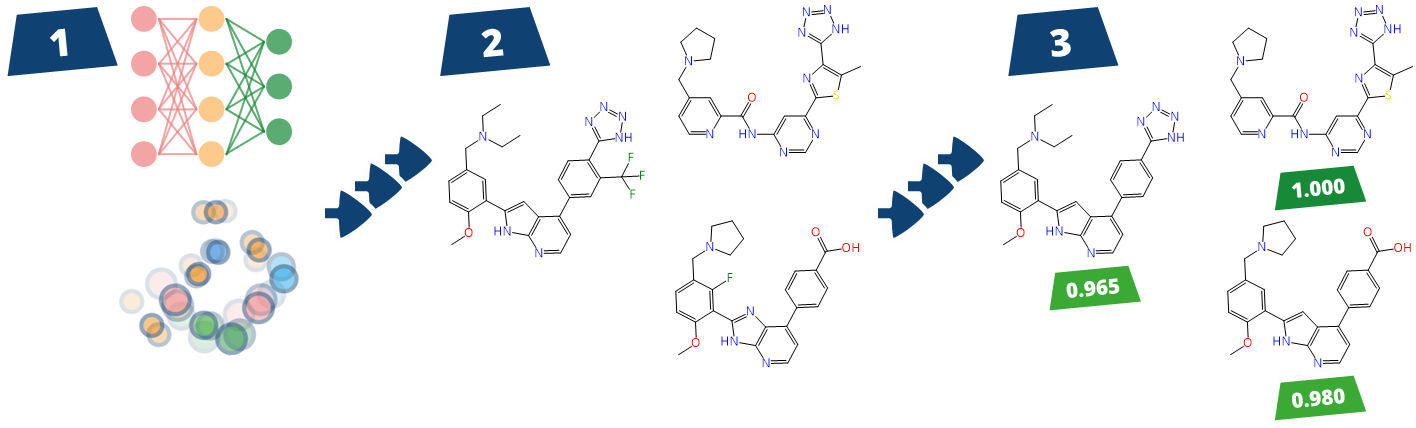
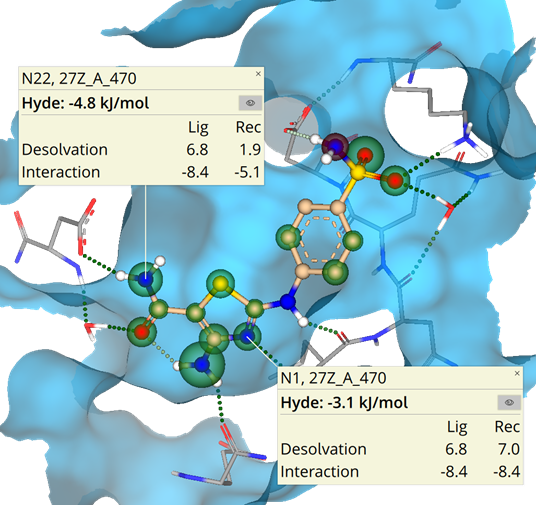
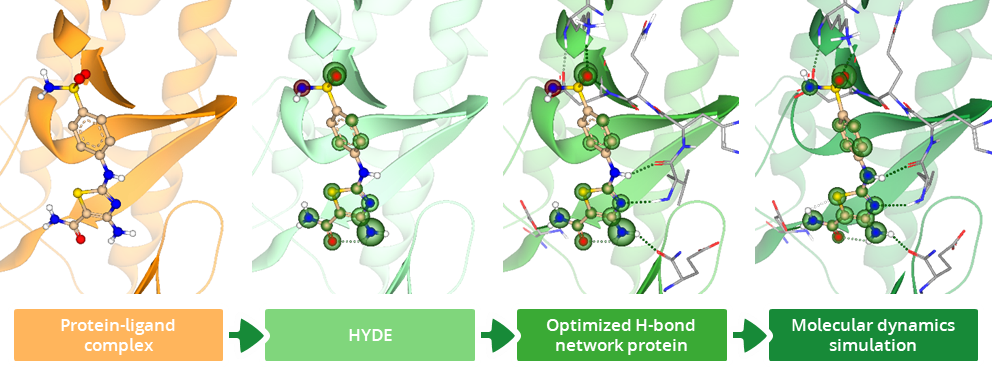
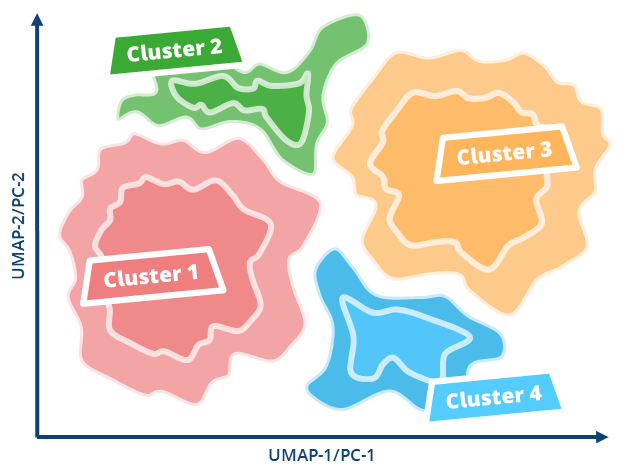
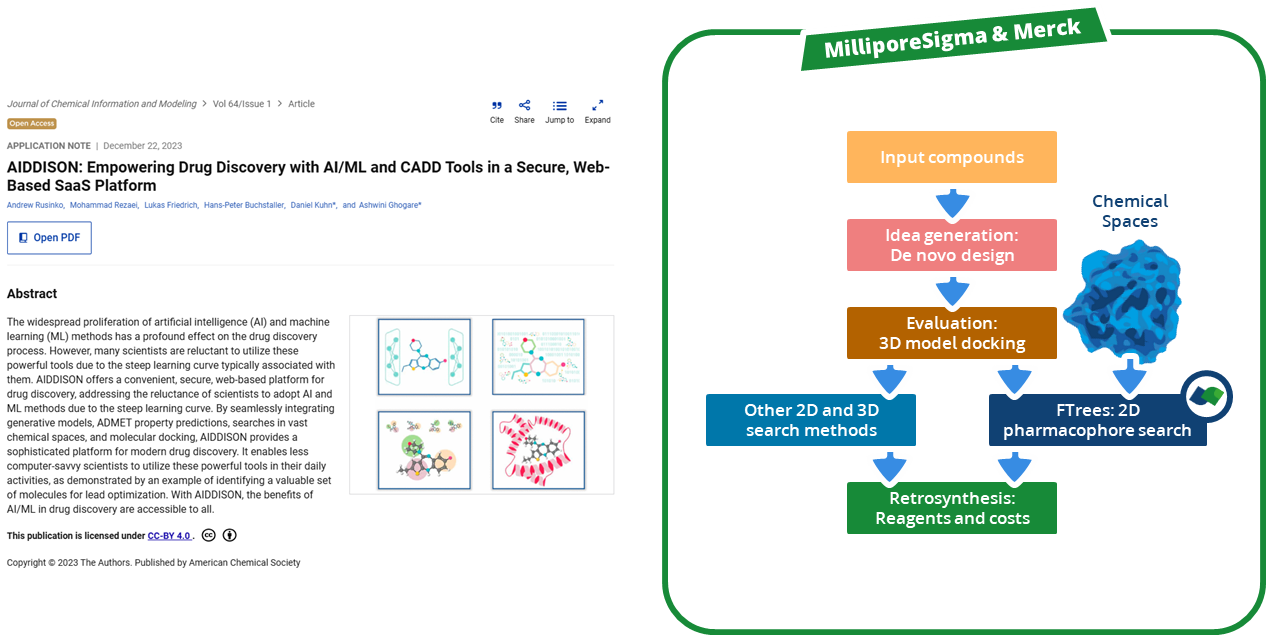
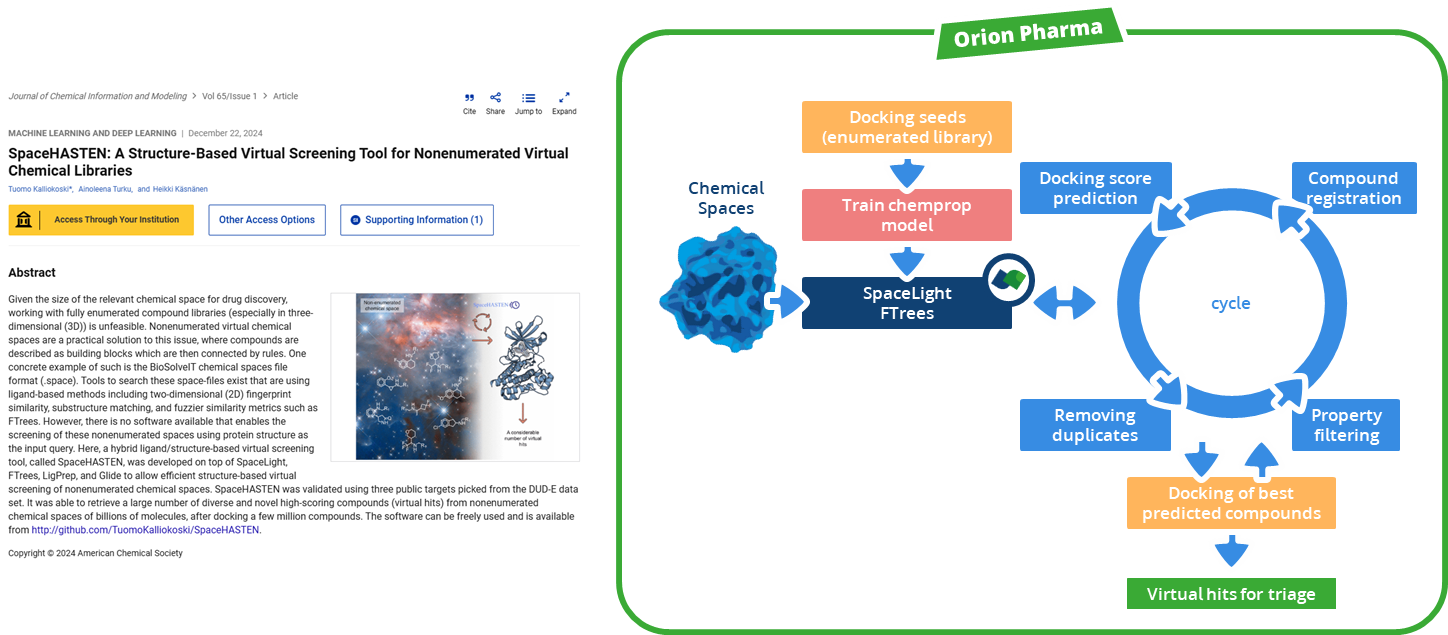
To enhance the virtual screening performance and compound selection,
Kuhn, Ghogare et al. from Merck and Millipore Sigma embedded Chemical Space navigation with FTrees into the AIDDISON workflow.
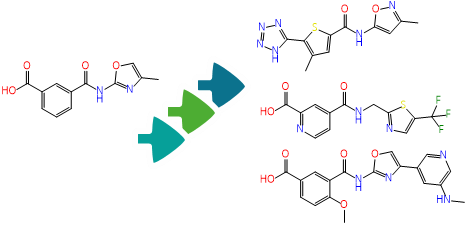
FTrees is used for 2D pharmacophore searches, enabling scaffold hopping and efficient ligand-based screening to expand the chemical diversity of related compounds to a proposed candidated. AIDDISON incorporates several Chemical Spaces, Enamine's REAL Space, WuXi's GalaXi, and their own in-house SA-Space, each covering billions of synthetically accessible molecules.
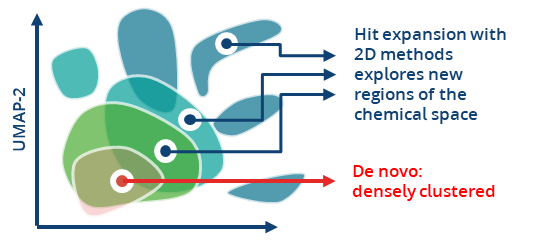
The integration of the search algorithm and the combinatorial Chemical Spaces accelerates searching, filtering, and identifying of promising drug-like molecules while expanding into uncharged areas of the chemical space that would not be covered by other 2D methods.
(Workflow representation modified after Kuhn, Ghogare et al. 2023.)