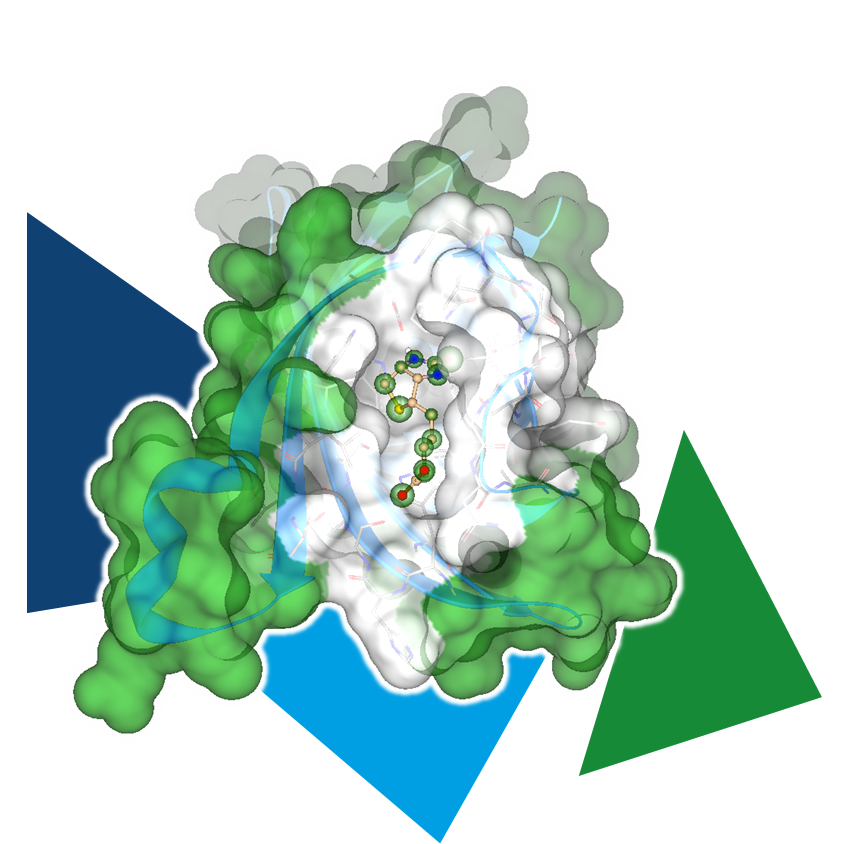
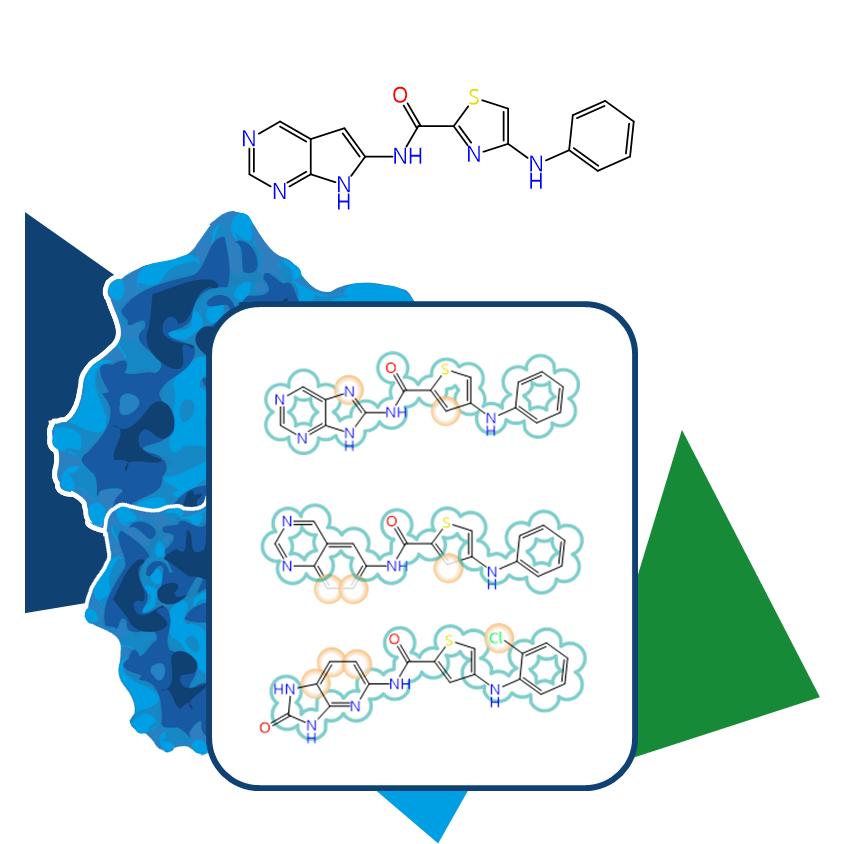
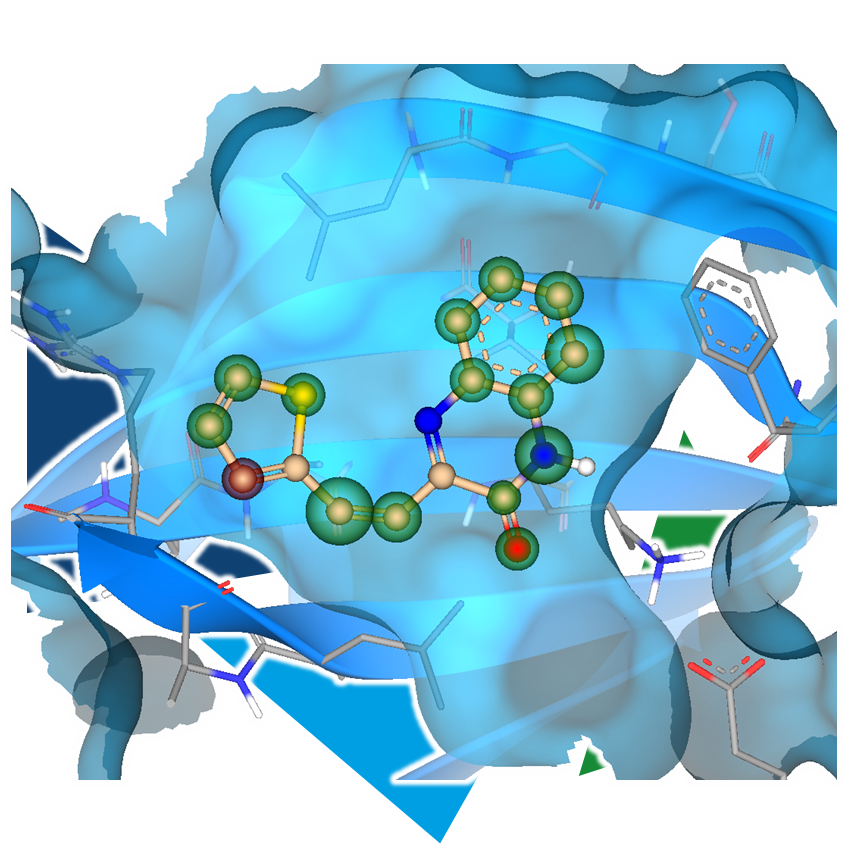
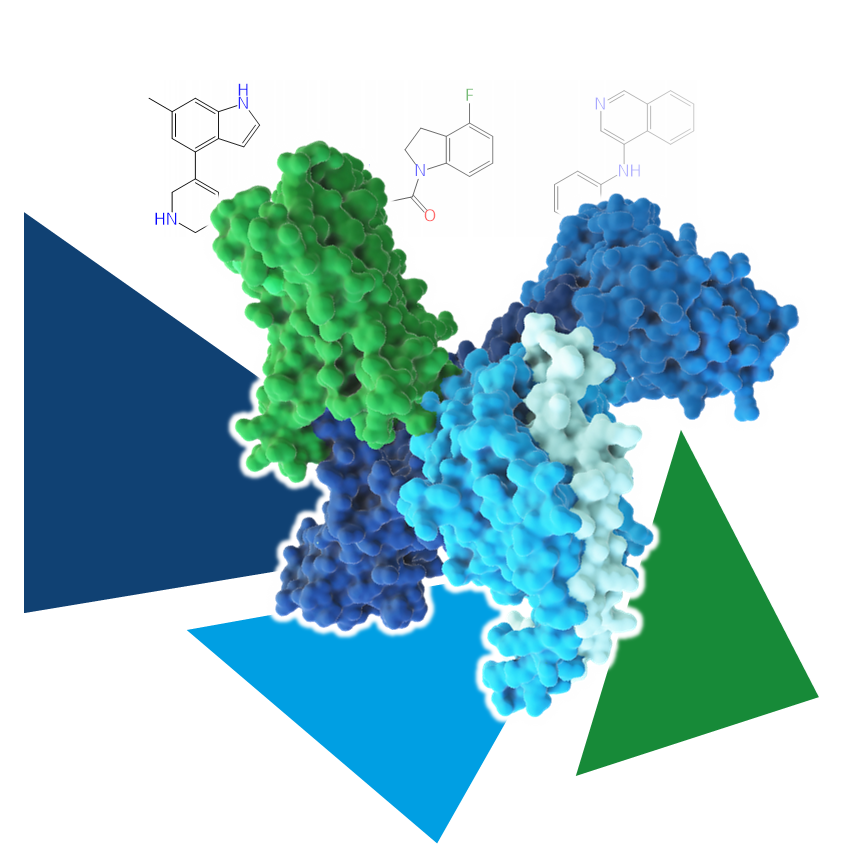
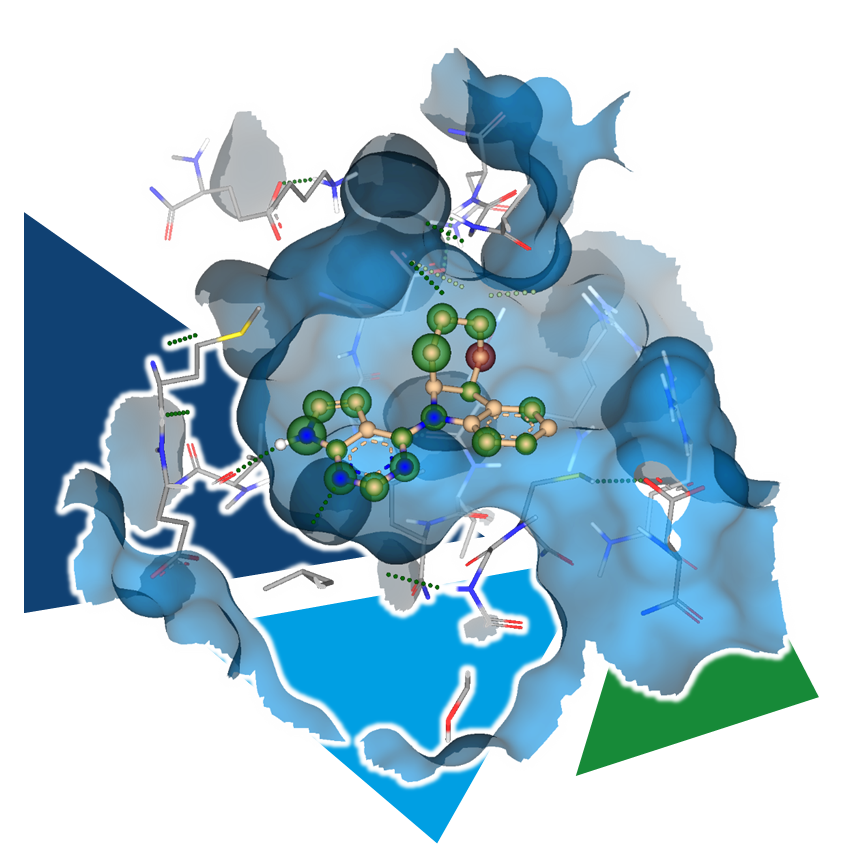
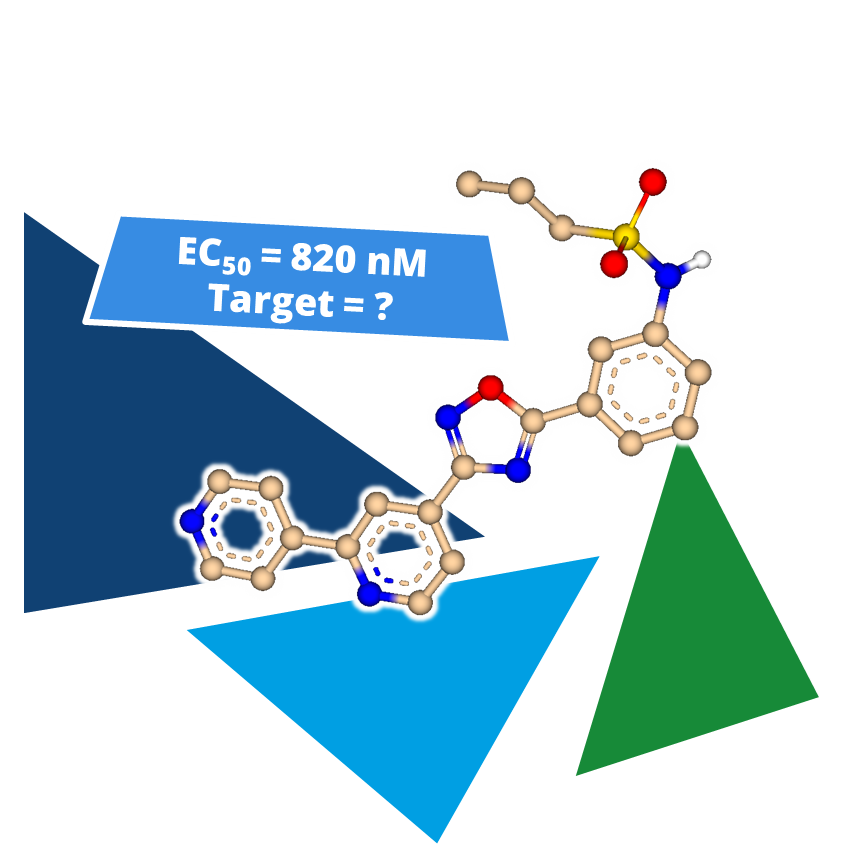
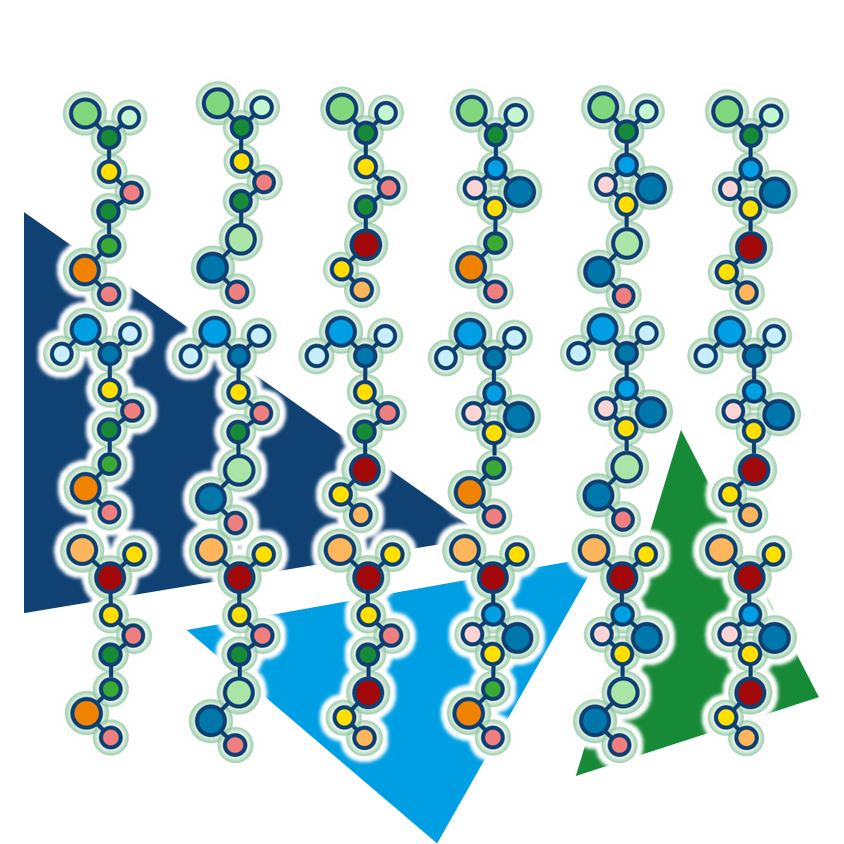
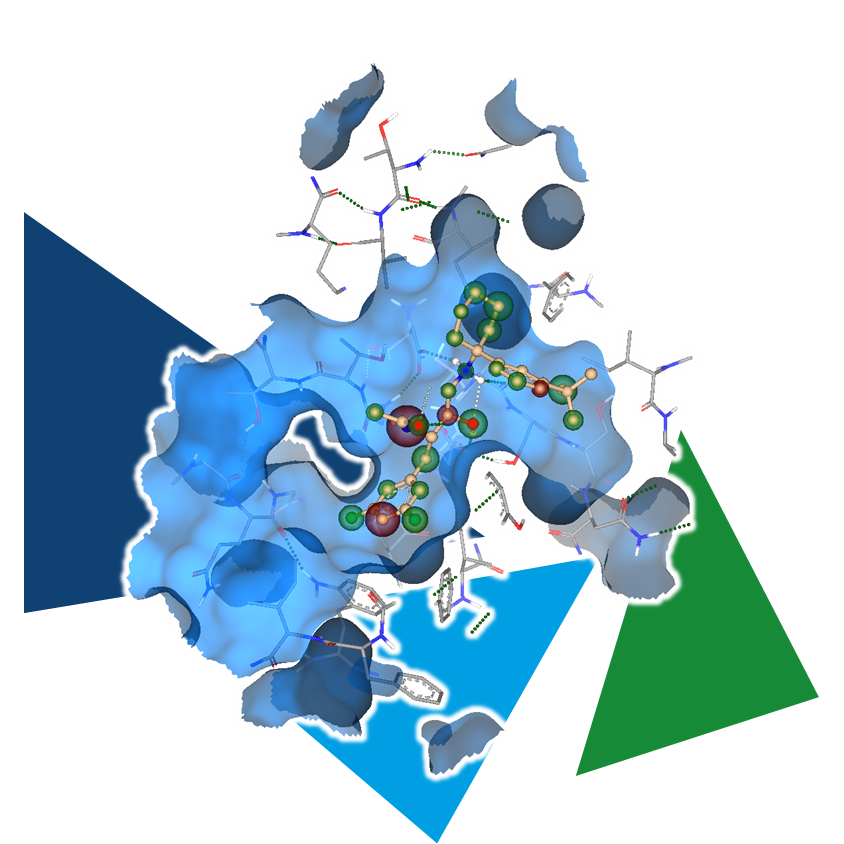
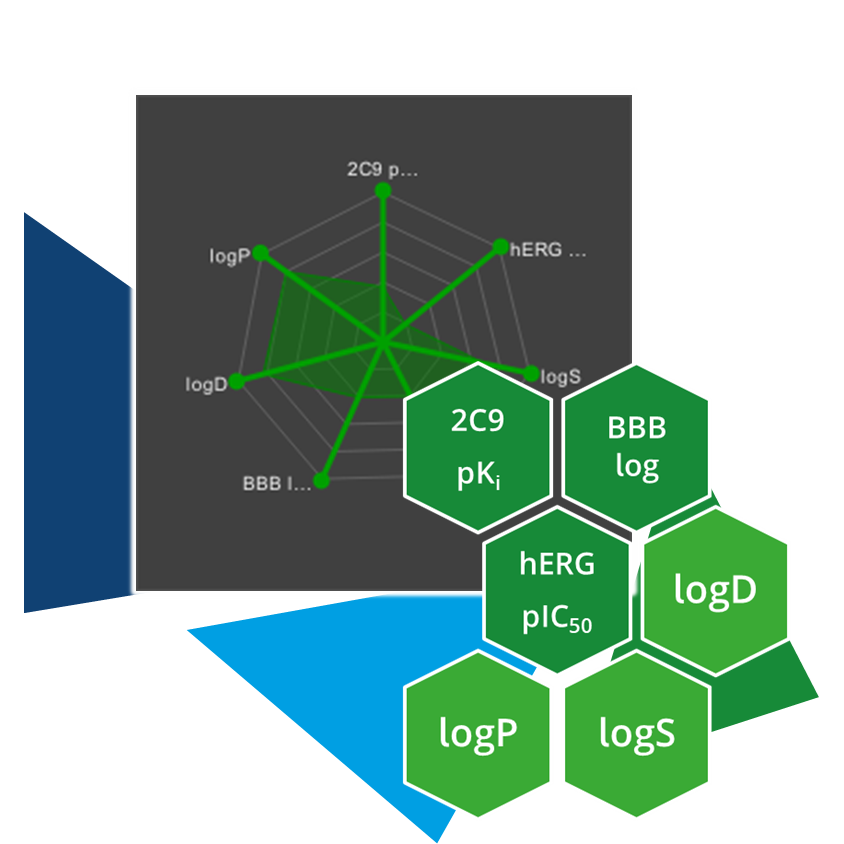
Even the most promising drug candidates can encounter roadblocks, including poor cell permeability, toxicity, and rapid metabolism, limiting their therapeutic potential.
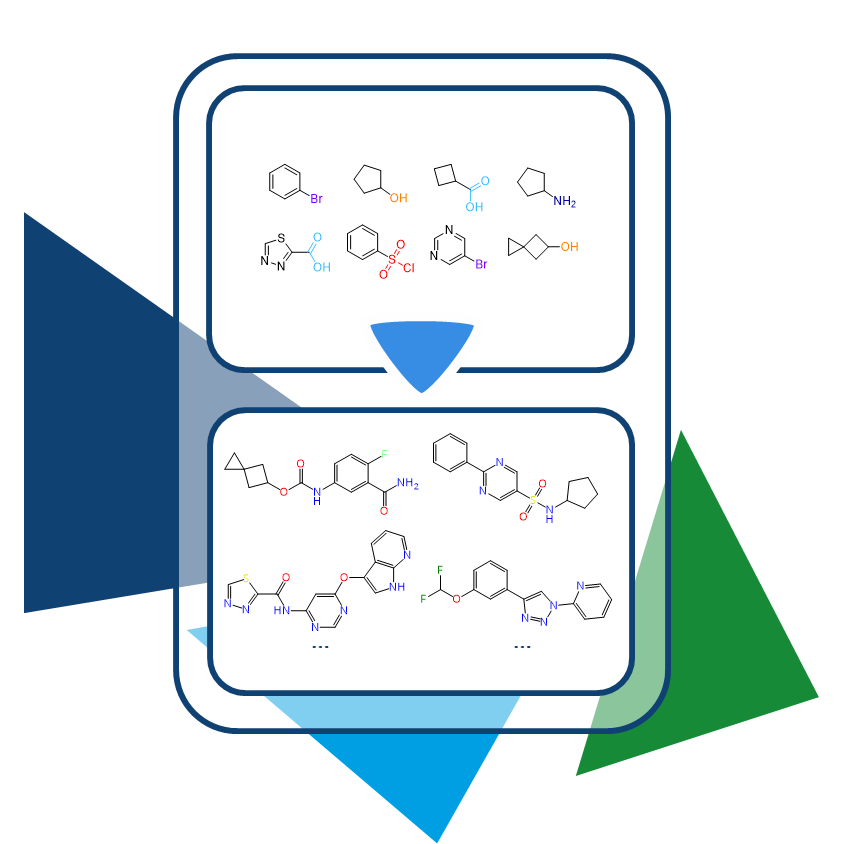
All the more frustrating when the target was promising from the start, and the compound itself had the potential to become a blockbuster. To maintain momentum, alternatives are needed—new molecules, ideally similar to what’s already there, just different. The same thing in another color, so to speak.
In the same vein, we shouldn’t forget that the strategy would be the same as when trying to circumvent the limitations around a blockbuster's patent...
To learn how to pull a miracle scaffold out of the hat, click
on this link.