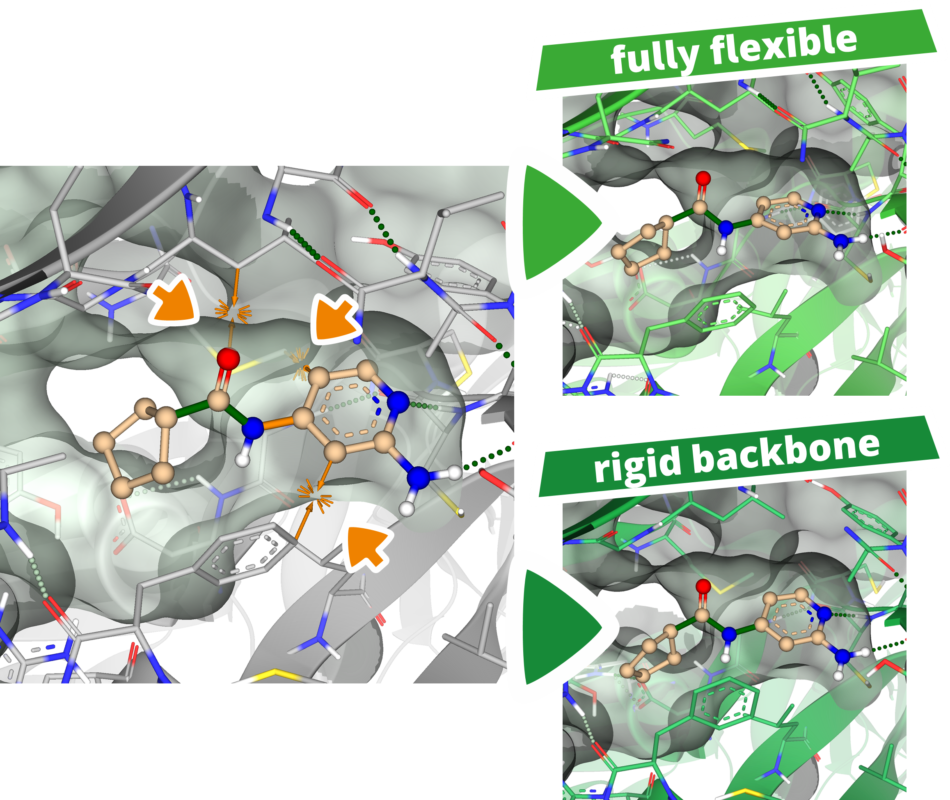
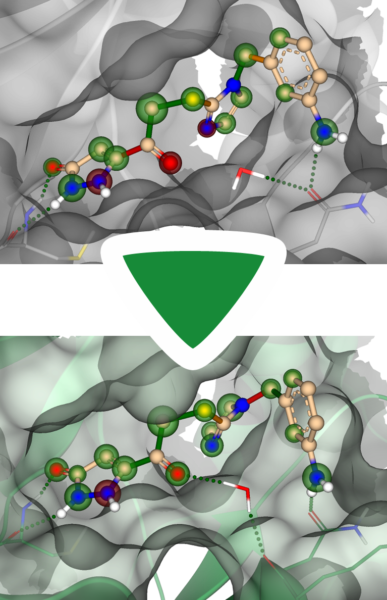
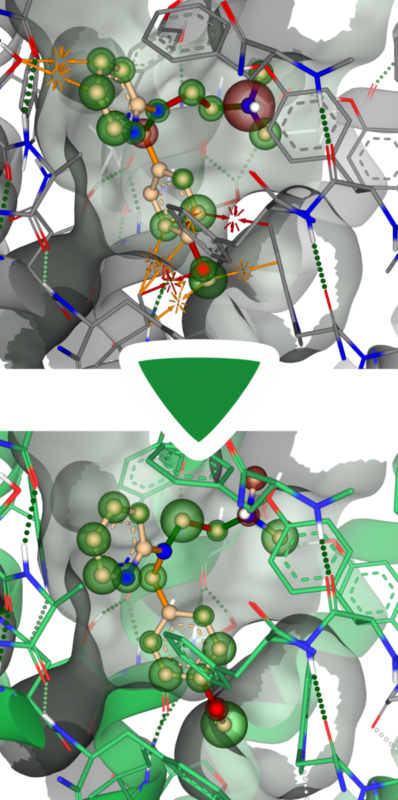
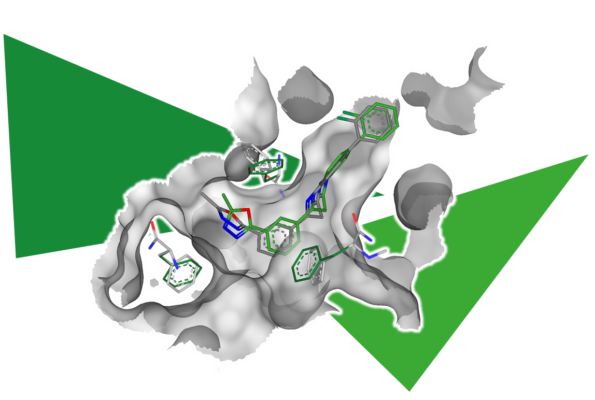
Energy minimization can also be utilized when the binding site of a structure is too small to host the ligand. Binding sites that are too narrow or virtually non-existent make docking predictions more challenging.
To simulate the induced fit of the target structure to the ligand, one can place the ligand in the binding site and temporarily tolerate clashes with the structure. By performing energy minimization afterward, both the ligand and the structure are allowed to adapt to each other (induced fit), thereby creating more space. In the example shown, clashes with the predicted model were successfully resolved after energy minimization.
Additional application scenarios:
- Generate space by expanding the ligand into potential subpockets.
- Overlay target subtypes or different X-ray structures to adapt a particular structure of interest to your ligand.
- Explore potential rotamers or combinations thereof that may expand the binding site size of your (apo) structures.
- Create space to host modalities to enable covalent docking.