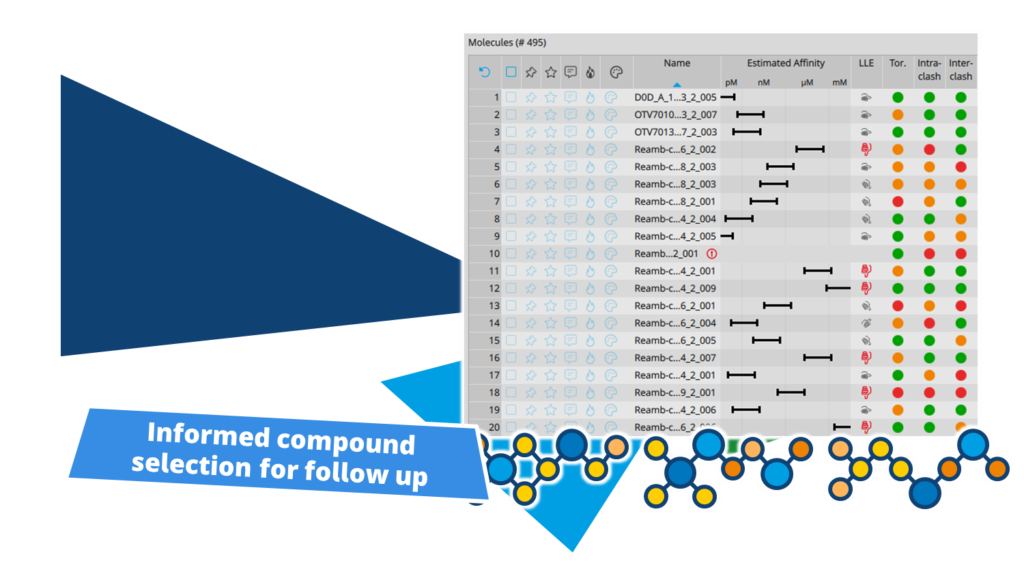
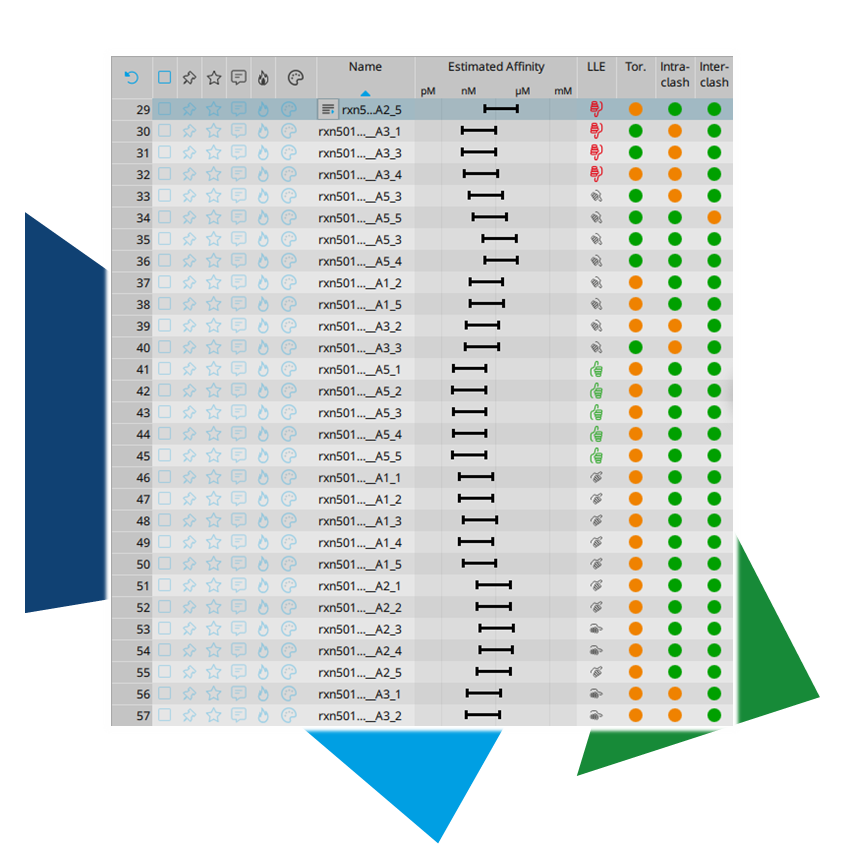
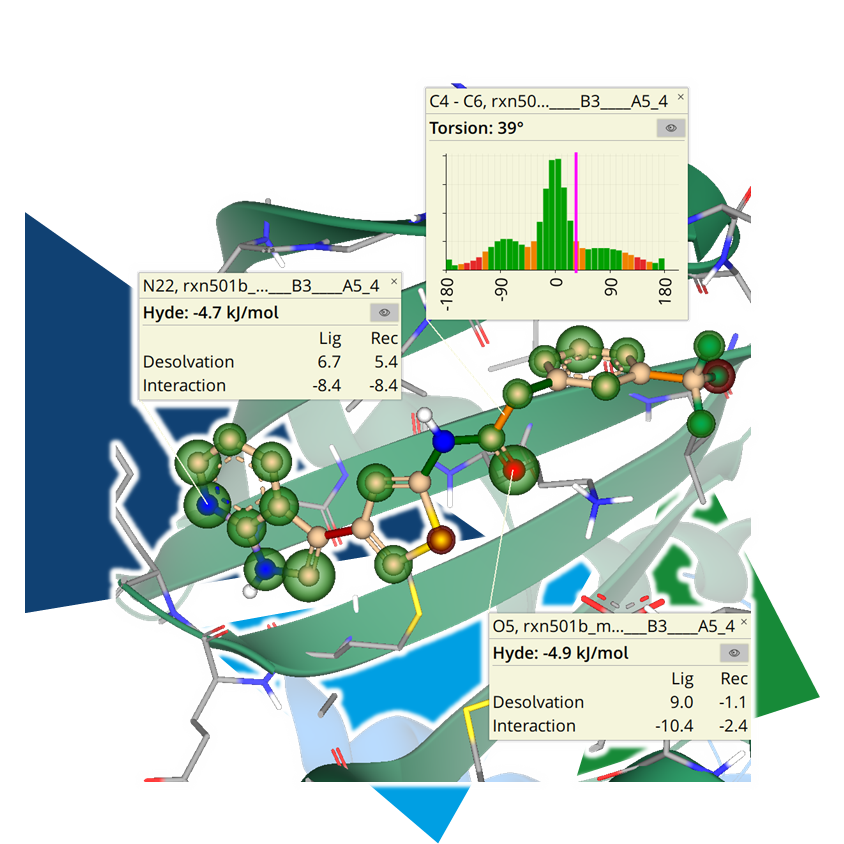
In partnership with
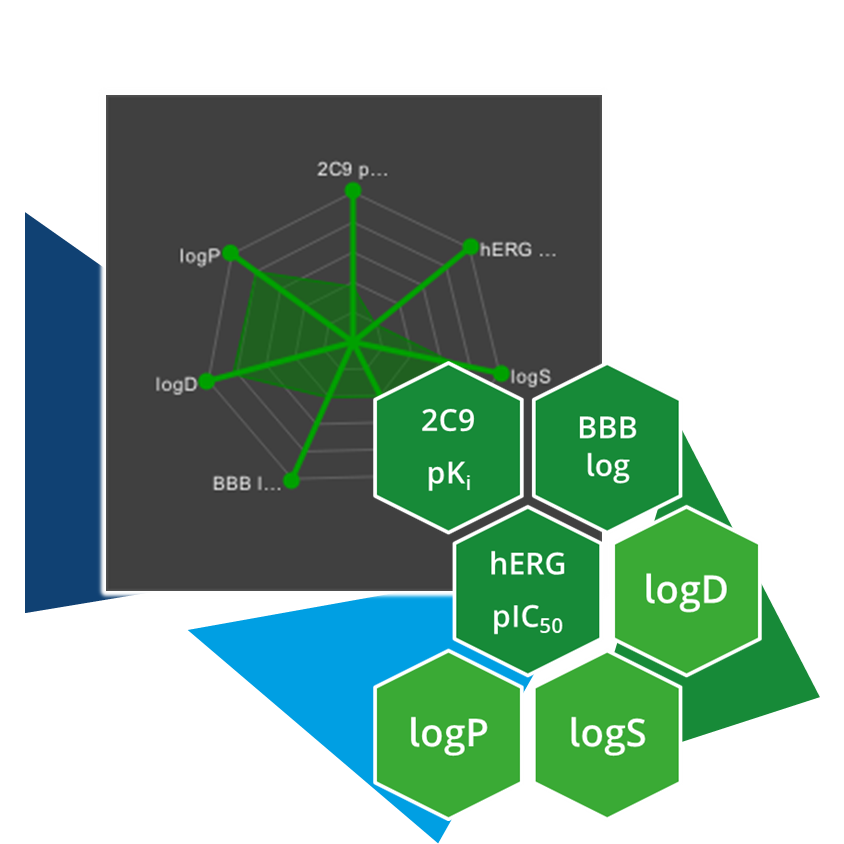
Optibrium, it is also possible to take it a step further and calculate ADME properties using the available models.
The models include: CYP2C9 pK
i, CYP2D6 affinity category, blood-brain barrier classification and CNS penetration, HIA category, P-gp category, PPB90 category, hERG pIC
50, logD, logP, logS, logS at pH 7.4.
Users can also import their own calculated models into our software to cover additional parameters.
Read more about Optibrium property prediction by
reading this PDF.