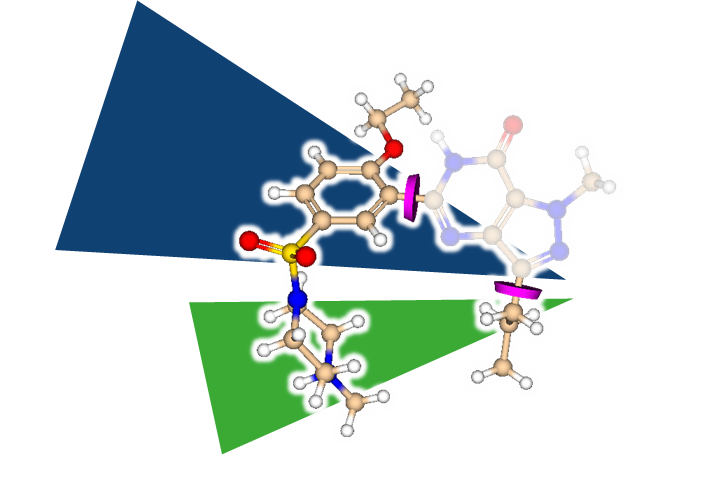
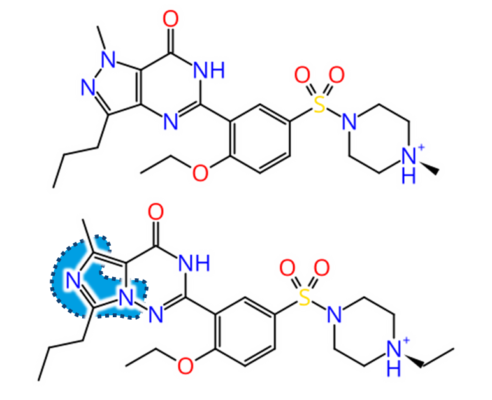
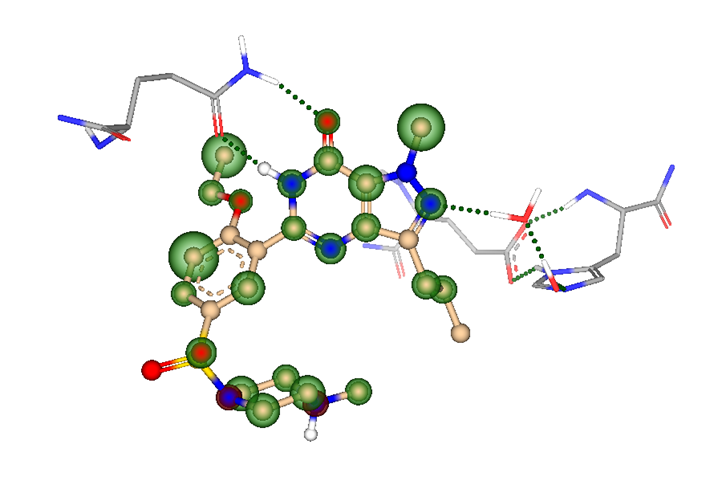
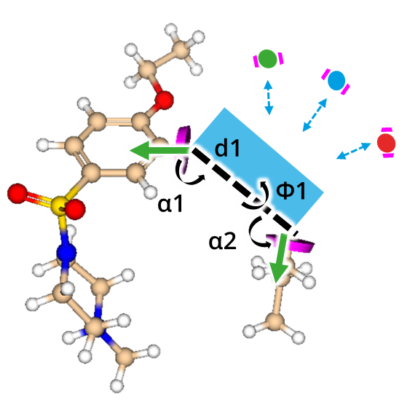
But how to discover replacements for a scaffold?
Virtual screening is a method to predict potential binders for a target of interest. A set of molecules is docked and scored allowing comparison of their predicted binding modes and interaction qualities. After selection of promising candidates, the compounds are aquired (through synthesis or external sources) and pharmacologically assessed. The greatest advantage of this method is that it can discover completely chemically unrelated candidates because it does not directly rely on structural information from a known binder.
Using pharmacophore contraints (like hydrogen bond acceptors/donors, lipophilic or charged groups, aromatic ring systems, etc.) can increase the success rate of the virtual screening as the generated poses will feature important interactions with the target.
Virtual screening
can be performed with SeeSAR.