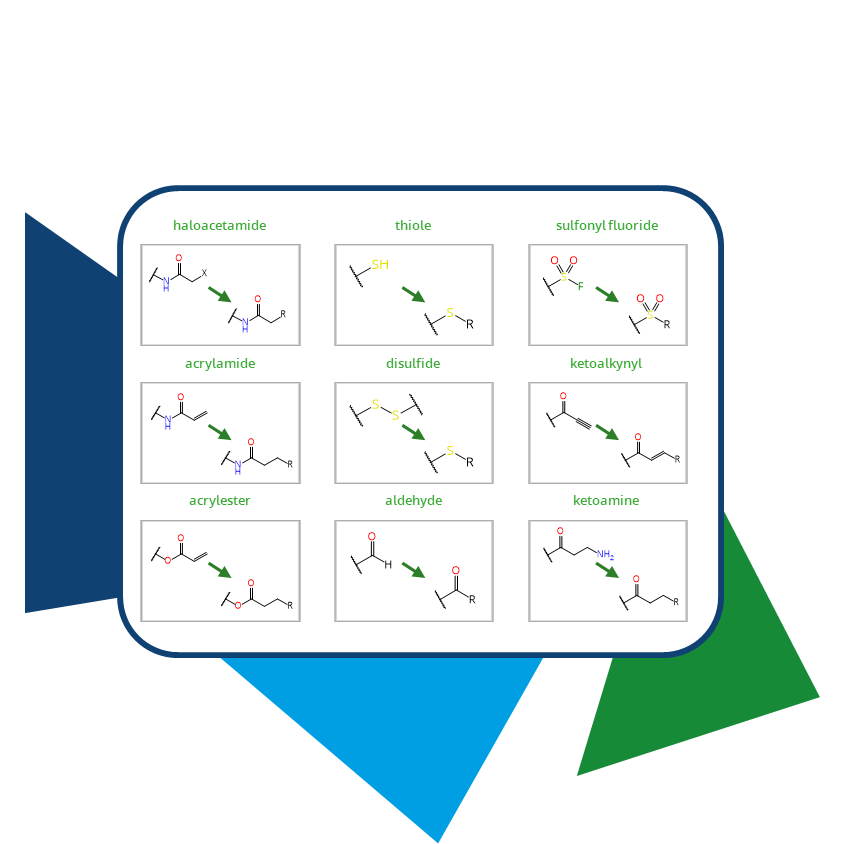
Moreover, Chemical Spaces are an excellent source of compounds containing warheads that are not found in standard catalogs. Their key advantage, beyond the novel intellectual property they represent, is their vast size. With billions or even trillions of entries, the number of compounds and specific warheads of interest increases significantly. Within our platforms infiniSee and infiniSee xREAL, it is possible to perform substructure searches for desired warheads. The command-line tool SpaceMACS further extends the search capabilities with SMARTS queries, enabling even more targeted searches for specific motifs.
BioSolveIT platforms for mining of covalent drug candidates:
- infiniSee: Chemical Space navigation platform with a graphical user interface.
- infiniSee xREAL: Exclusive platform to screen Enamine's largest compound catalog featuring trillions of compounds.
Command-line tools for mining of covalent drug candidates:
- SpaceMACS: Performs maximum common substructure searches, as well as exact substructure mining. Algorithm behind the Motif Matcher Mode.