With the recent advancements in AI technology, it has become prominent to apply machine learning approaches to in-house data sets in order to predict promising drug candidates with improved properties. A small drawback, however, is that even the most promising candidates may hold limited value if their synthesis is too complex or costly, potentially slowing down the entire R&D process. Unfortunately, integrating the synthesizability of a compound into a workflow designed to predict the best candidates is not a trivial task.
Here, we would like to focus on the options available to streamline the process and quickly reach the desired goals.
Leverage the Advantages of the Numbers Game
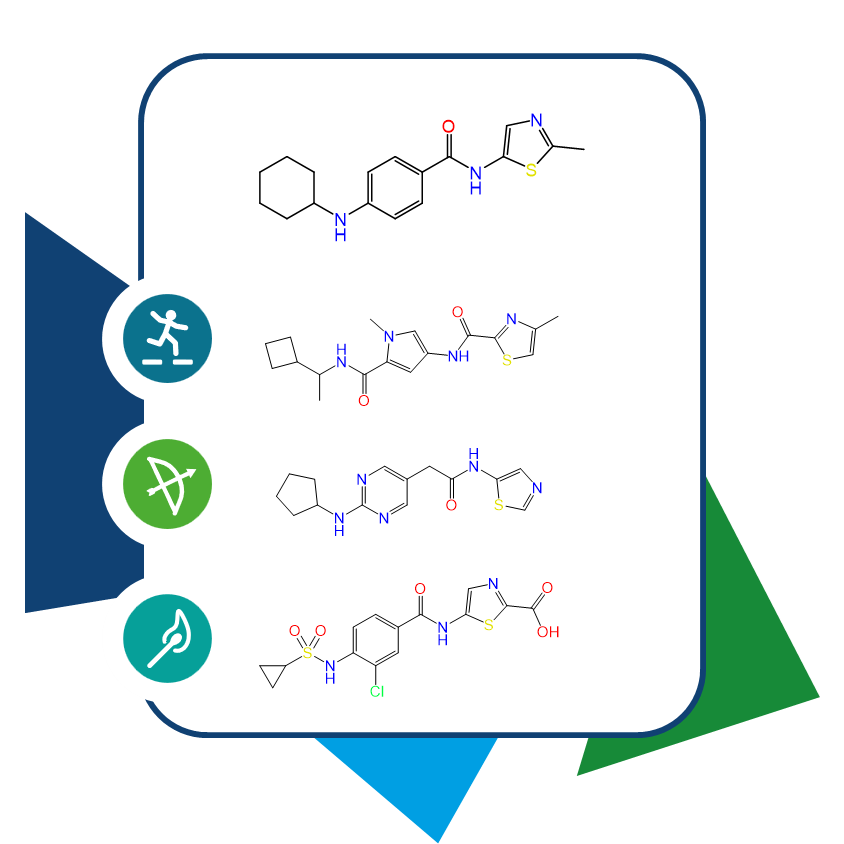
The largest problem with de novo compound design is the accessibility of the results. For a suggestion from a model, seven or eight synthesis steps are too much effort, which makes it highly unlikely that the compound will ever be synthesized. This is especially true in the early stages, where new chemotypes are being sought.
Therefore, uncomplicated access to the physical compound is the rate-limiting step, which ultimately determines the usability of the de novo method.
Modern medicinal chemistry can indeed be complex, but many will be surprised at how much of chemical space can be covered with just a few common reactions. With each additional reaction and the corresponding building blocks, one quickly enters the billions or even trillions. This is where the numbers game begins: through ultra-large numbers, one can come very close to the results proposed by machine learning methods or even find them exactly. The approach to the chemical space around a compound already uncovers additional analogs, which can also be considered alternatives for testing.
Looking at De Novo Compounds from Different Angles
What’s interesting is that the approach to the 'most similar' compounds can take place from various angles. For example, it’s possible to search for compounds that share functional groups with a template molecule, those with the highest molecular fingerprint similarity, or those that share the largest sequence of heavy atoms. The question can change depending on the project or compound class, so having a selection of different screening methods is advantageous to act accordingly. These methods can also be used orthogonally to find a similarity consensus or chained together to search for compounds that are considered similar to the query from all perspectives.
BioSolveIT software for mining similar compounds from Chemical Spaces:
infiniSee: Chemical Space navigation platform with a graphical user interface.
infiniSee retrieves relevant chemistry from ultra-large Chemical Spaces containing billions or even trillions of compounds based on their similarity to a query compound. Results are synthetically accessible per design in one or two steps and, in the case of our partners' Chemical Spaces, can be ordered directly to your table.
infiniSee xREAL: Exclusive platform to screen Enamine's largest compound catalog featuring trillions of compounds.
infiniSee xREAL contains all features of infiniSee and supports all three Chemical Space exploration search modes.
Command-line tools for mining similar compounds from Chemical Spaces
FTrees: Pharmacophore-based similarity screen. Algorithm behind the Scaffold Hopper Mode.
SpaceLight: Retrieves close analogs based on molecular fingerprints. Algorithm behind the Analog Hunter Mode.
SpaceMACS: Performs maximum common substructure searches, as well as exact substructure mining. Algorithm behind the Motif Matcher Mode.
Efficient Accessibility
Aside from their vast size, Chemical Spaces offer additional advantages that make them particularly attractive. The real game-changer—and the primary reason they are highly valued in the pharmaceutical industry—is that the resulting compounds can be directly ordered from partner compound providers who have set up their own Chemical Spaces.
With competitive pricing, these providers offer a cost-effective way to access desired compounds or significantly reduce the burden on in-house synthesis teams. This allows researchers to quickly obtain in vitro results without the time-consuming and resource-intensive process of custom synthesis, thus accelerating early-stage drug discovery efforts.
Translating into Tangability in Action
In this publication, authors from MilliporeSigma specifically address the question of how to efficiently integrate de novo design into a drug discovery workflow. For the setup of their Chemical Spaces (SA-Space) and its screenings, they use BioSolveIT technologies CoLibri and FTrees.
The approach demonstrates how machine learning methods can be used to eventually lead to tangible compounds.