Manually editing a structure is certainly possible for a small number of analogs to explore the effects on binding affinity. However, if you want to explore the chemical space around the compound more efficiently, you can use another tool.
In SeeSAR's
Inspirator Mode, there is the 'Analoging' feature, which uses the
MedChemesis engine: MedChemesis is a ligand mutation tool that takes the starting molecule and applies 290 of the most commonly used MedChem transformations to look for potentially improved analogs. The transformations include adding functional groups and heavy atoms, exchanging individual atoms in a chain or ring system, as well as converting motifs into their bioisosteric counterparts.
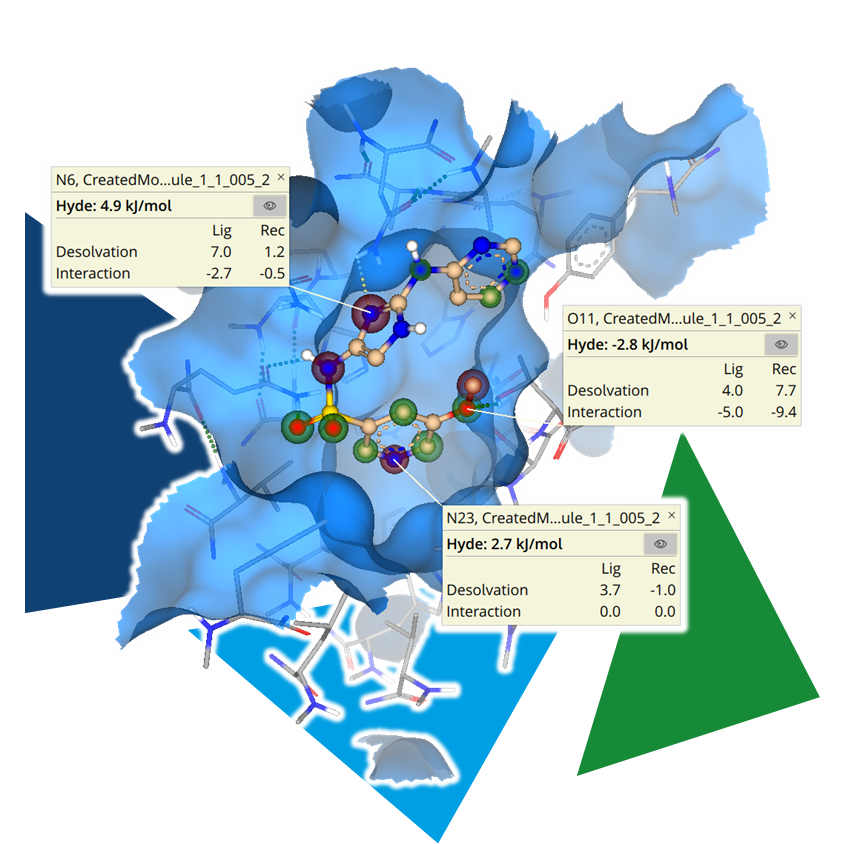
The fine-grained exploration of the binding site allows, among other things, the identification of positions for growth, as well as potential sites for H-bond interactions. Furthermore, it enables the generation of ideas for where lipophilic groups can be tolerated or even increase the ligand's affinity.