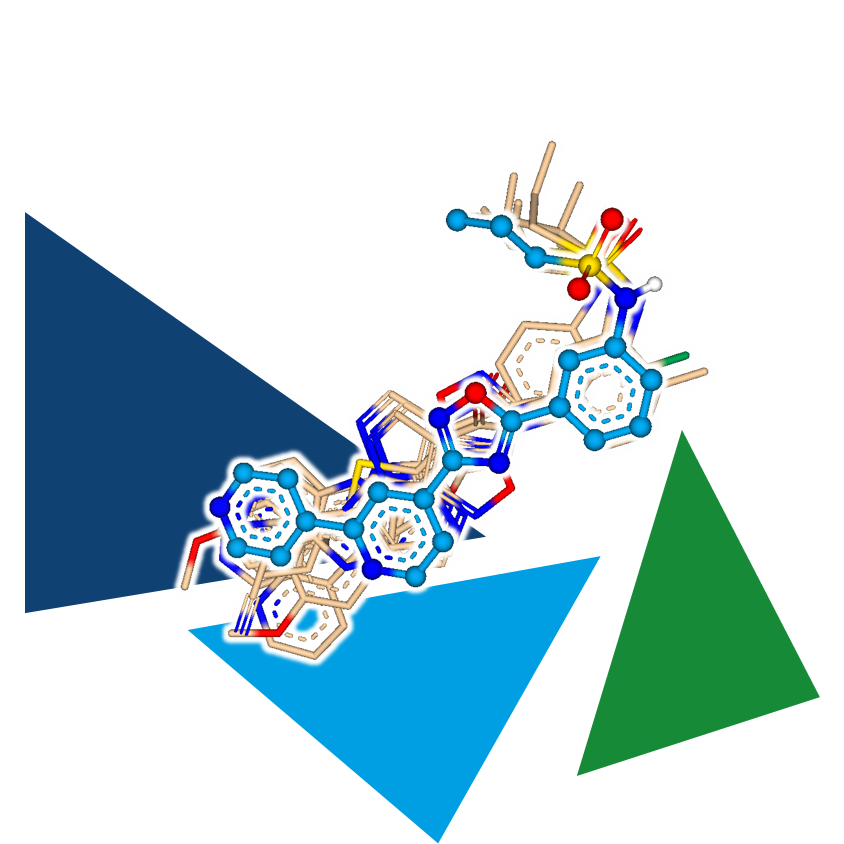
2D methods have the disadvantage that by reducing information to the 2D world, some data points are lost, which can significantly decrease the hit rate. Therefore, it may be beneficial to combine them with other methods to achieve hit enrichment. For example, after performing a fuzzy pharmacophore screening, one could continue working with the results in the 3D world. By using a known binder as a template and subsequently superposing the molecules in 3D, a second virtual screening filter can be applied to enrich promising candidates that resemble active molecules in 3D.
BioSolveIT software for 3D alignment:
- SeeSAR's Similarity Scanner Mode: Ligand-based virtual screening.
Command-line tools for 3D alignment:
Of course, it is a very good idea to extract a large, but still manageable number of molecules from a Chemical Space, and then, if possible, dock them at the target. This method has the advantage of requiring docking of a significantly smaller number of molecules from billions or trillions, which keeps the required computational resources lean.