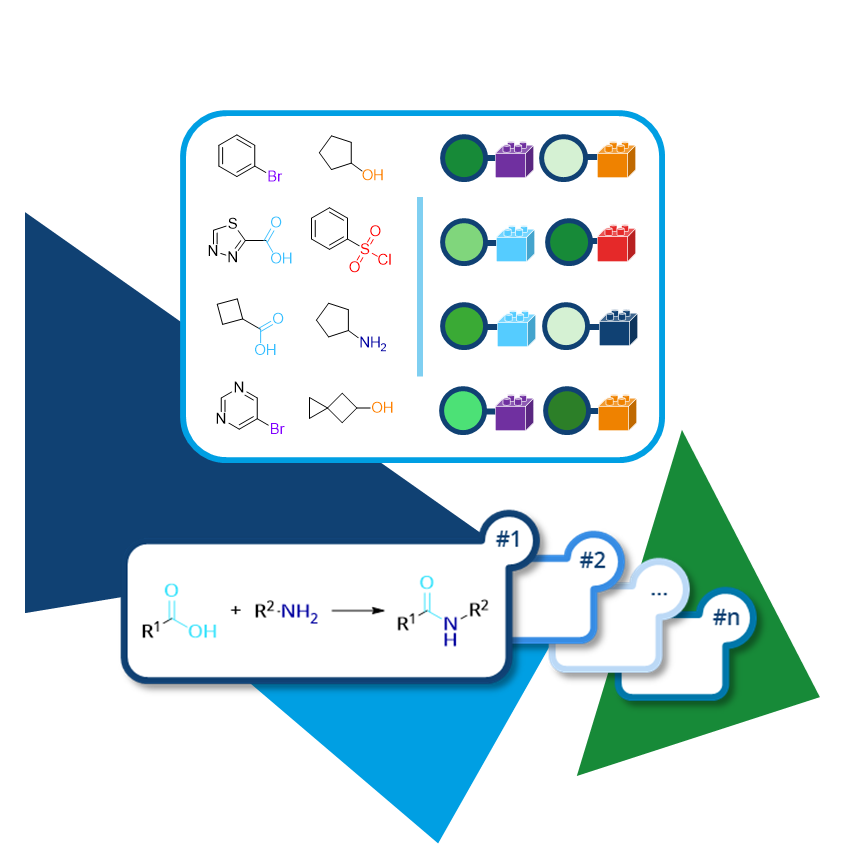
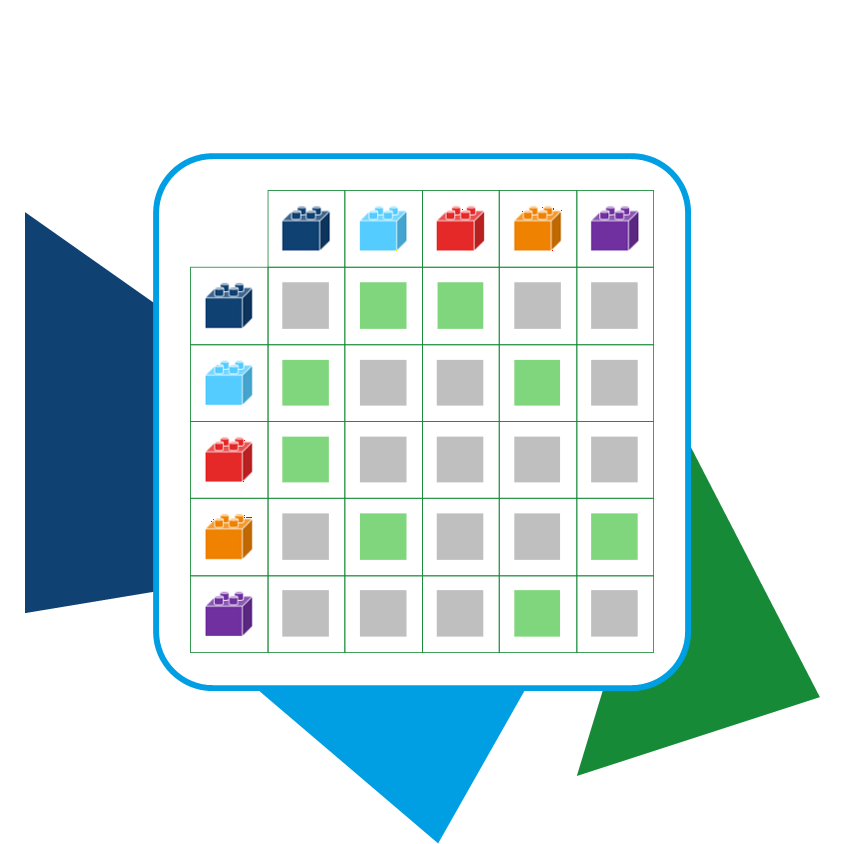
The huge numbers of Chemical Spaces are the result of a combinatorial explosion that occurs when the building blocks are combined through
predefined reactions. A combination of 1,000×1,000×1,000 building blocks already results in 10
9 compounds. Therefore, Chemical Space scale exponentially with their building blocks and included reactions.
The chemical reactions have to be written in the
SMIRKS format to define the pattern how the building blocks are combined.
You can use a collection of robust, chemical reactions that was used in the creation of
eMolecules eXplore Chemical Space that is
available as download here as a starting point for the setup of your Space.
Again, the real value is added by adding your own in-house reactions to cover a larger area of unexplored chemical entities.