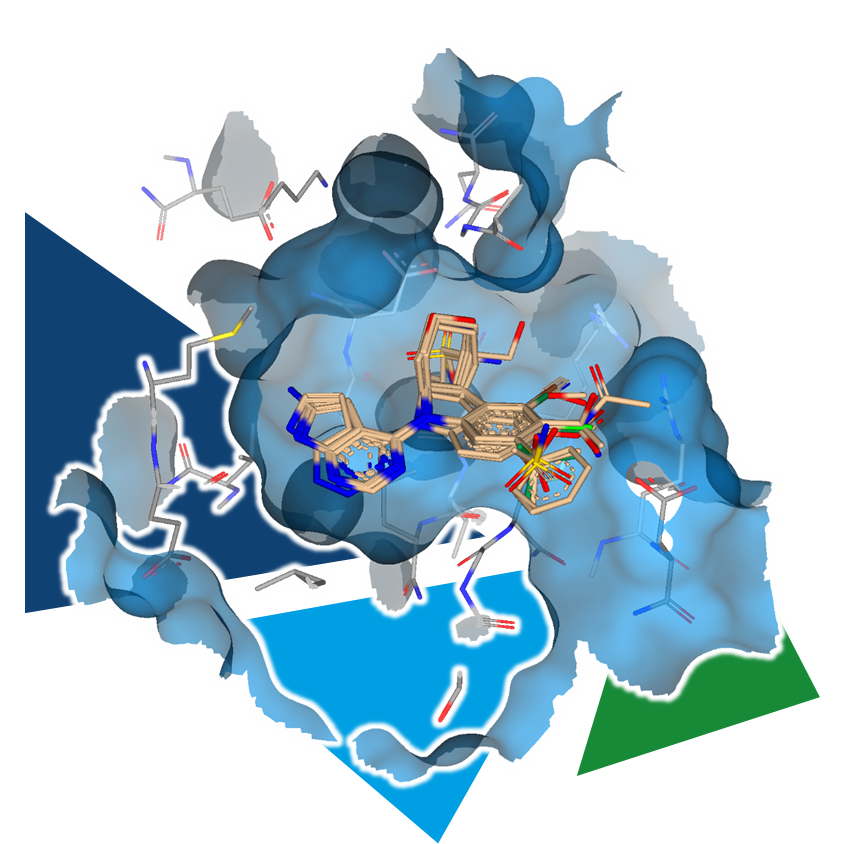
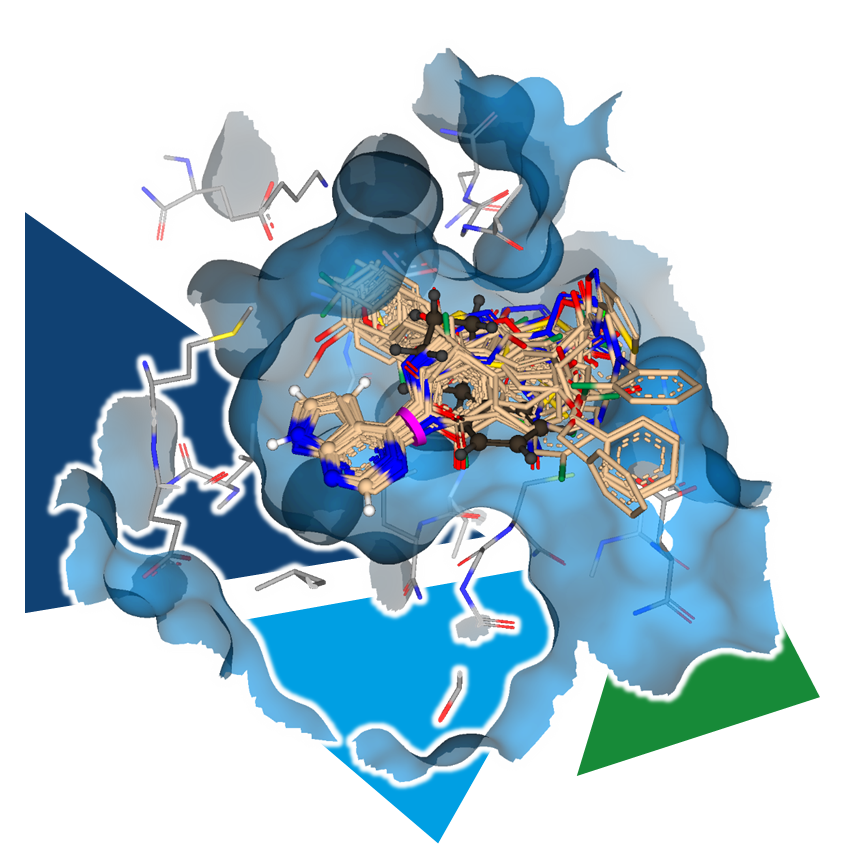
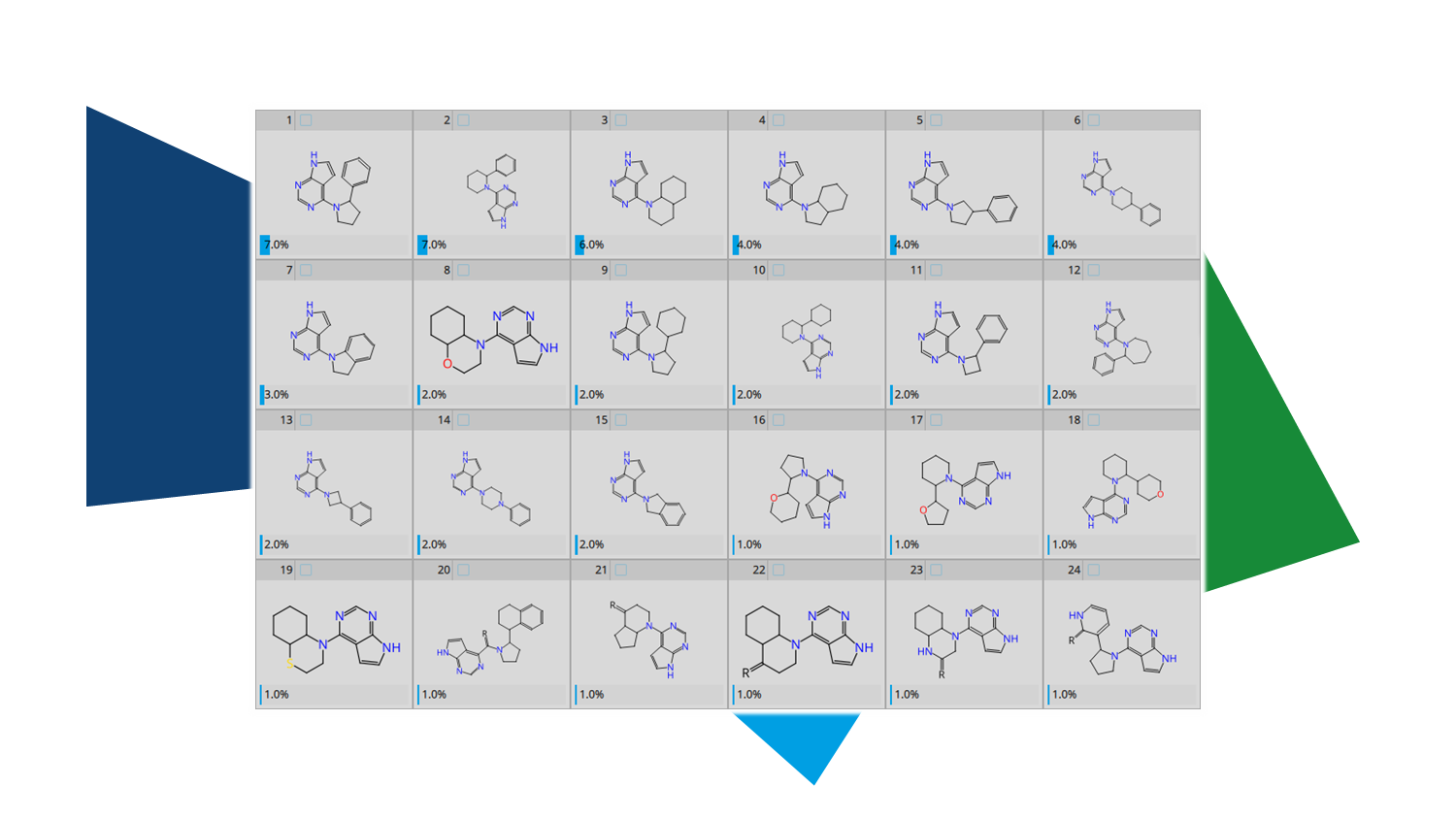
The lack of information on the target's structure or uncertainties about the binding mode of the ligand can impede structure-based drug design mentioned above.
In those cases, ligand-based approaches bypass the 3D requirements and enable focused lead optimization.
By systematically purchasing or synthesizing analogs of a compound with varying decorations and subsequently assessing their biological activity, SARs can be established step by step. The commercial availability of these analogs determines how quickly insights can be gained. The more relevant analogs available, the faster the series can be explored and developed. Accordingly, the potential of a compound collections increases with its size and comprised analogs to a molecule of interest.
Chemical Spaces are ultra-large compound collections of accessible compounds. Taking the "SAR by catalogue" concept into the Chemical Space world, "
SAR by Space" emerges as an efficient method to profit off the huge numbers of commercially available compounds.