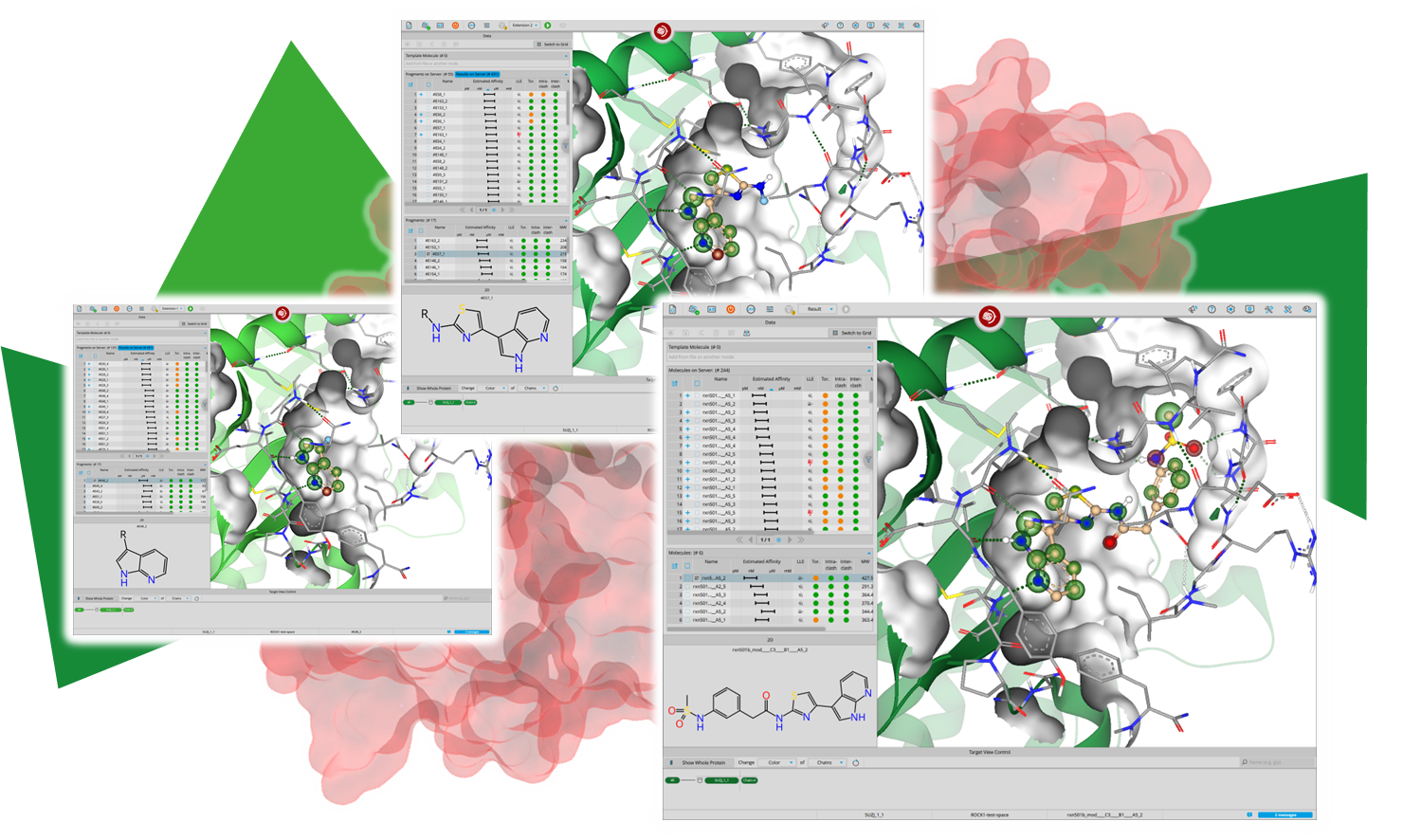
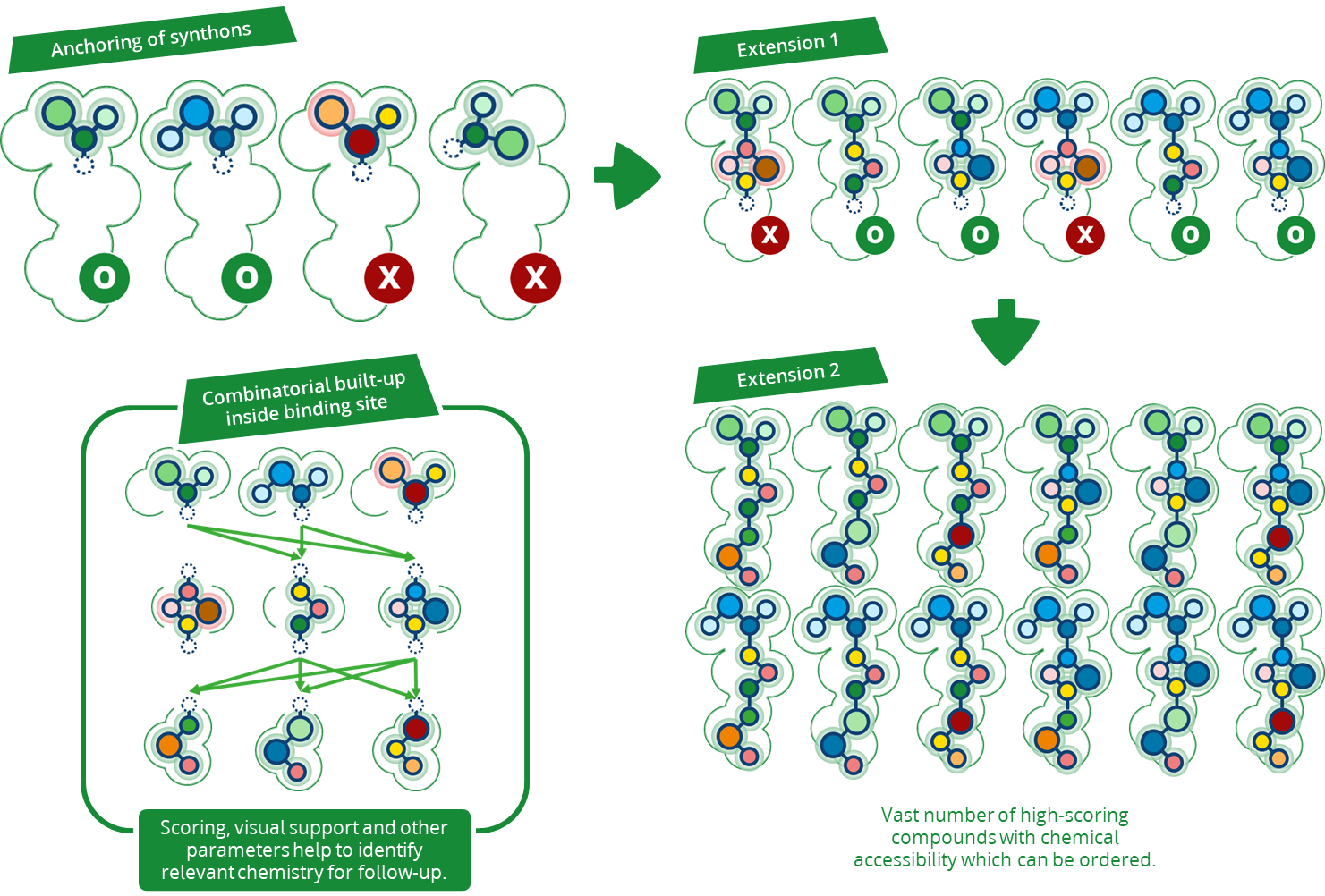
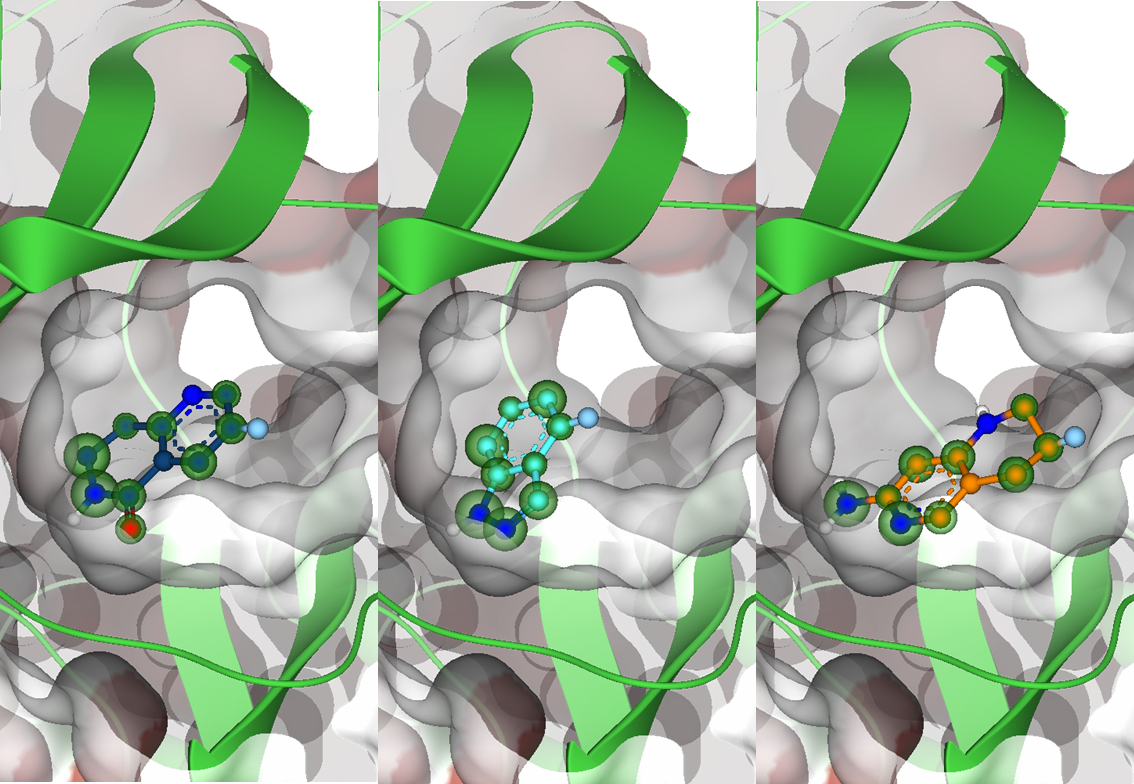
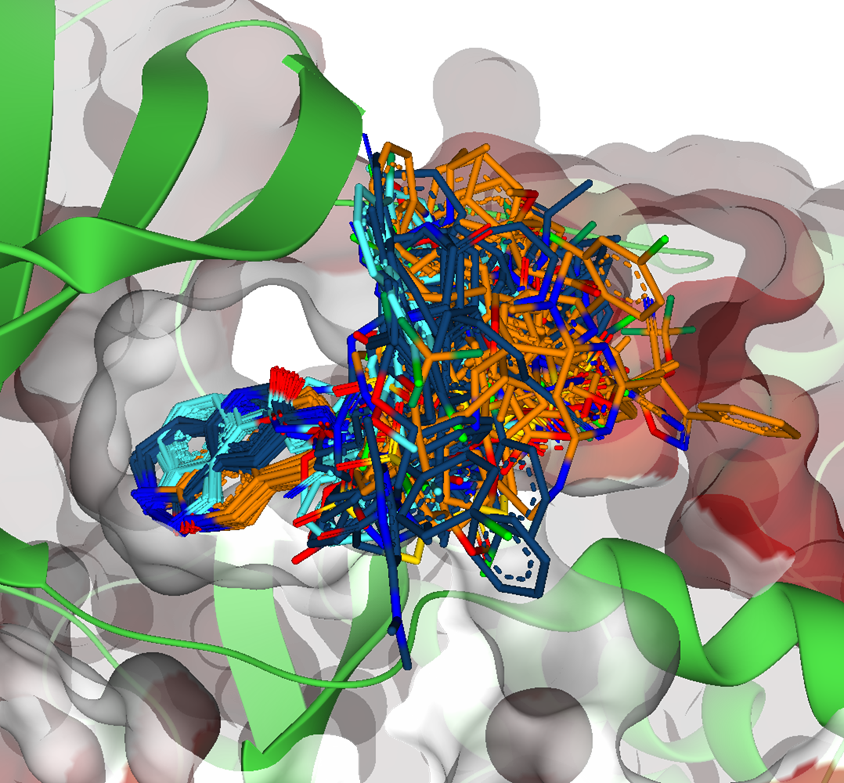
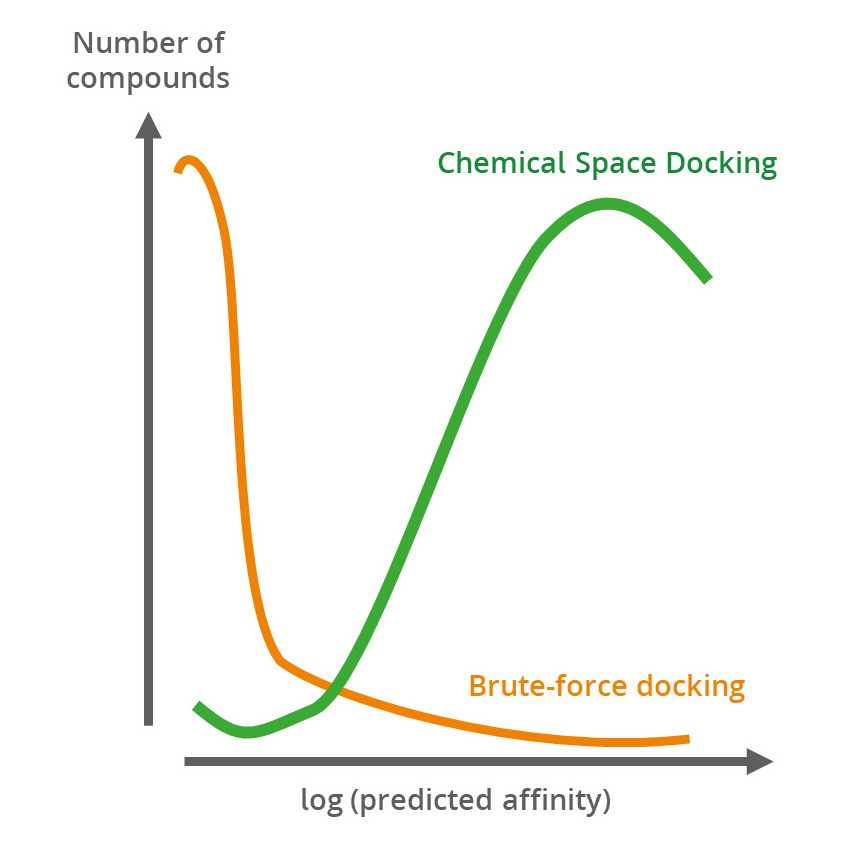
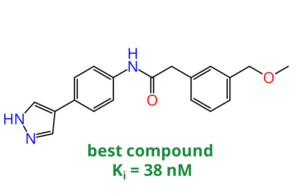
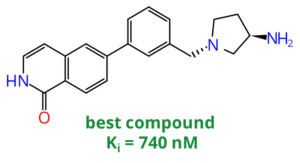
Magnet for the Needle in Haystack: “Crystal Structure First” Fragment Hits Unlock Active Chemical Matter Using Targeted Exploration of Vast Chemical Spaces.
Müller, J.; Klein, R.; Tarkhanova, O.; Gryniukova, A.; Borysko, P.; Merkl, S.; Ruf, M.; Neumann, A.; Gastreich, M.; Moroz, Y.S.; Klebe, G.; Glinca, S.
Journal of Medicinal Chemistry 2022, 65 (23), 15663-15678.